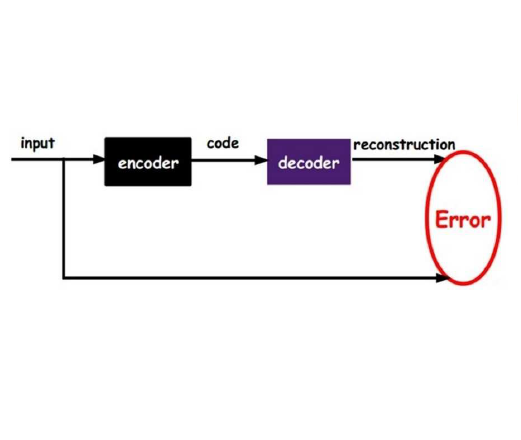
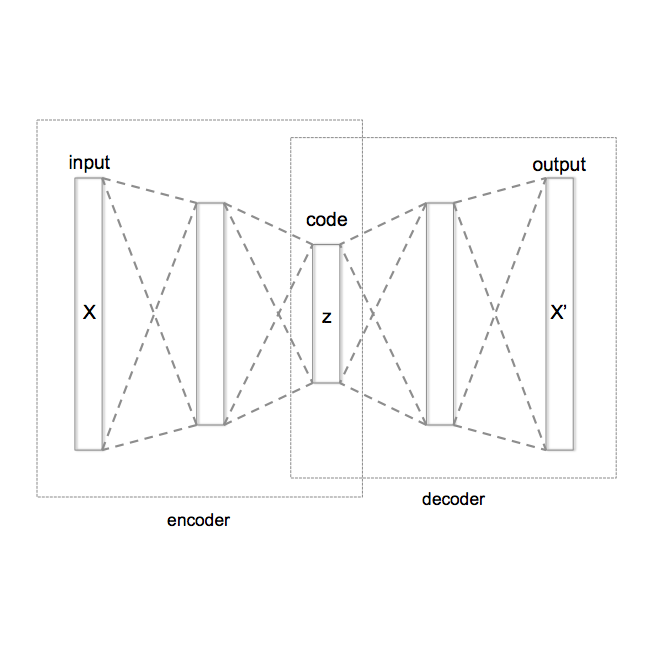
Cryo-electron microscopy (cryo-EM) is capable of producing reconstructed 3D images of biomolecules at near-atomic resolution. As such, it represents one of the most promising imaging techniques in structural biology. However, raw cryo-EM images are only highly corrupted - noisy and band-pass filtered - 2D projections of the target 3D biomolecules. Reconstructing the 3D molecular shape starts with the removal of image outliers, the estimation of the orientation of the biomolecule that has produced the given 2D image, and the estimation of camera parameters to correct for intensity defects. Current techniques performing these tasks are often computationally expensive, while the dataset sizes keep growing. There is a need for next-generation algorithms that preserve accuracy while improving speed and scalability. In this paper, we combine variational autoencoders (VAEs) and generative adversarial networks (GANs) to learn a low-dimensional latent representation of cryo-EM images. We perform an exploratory analysis of the obtained latent space, that is shown to have a structure of "orbits", in the sense of Lie group theory, consistent with the acquisition procedure of cryo-EM images. This analysis leads us to design an estimation method for orientation and camera parameters of single-particle cryo-EM images, together with an outliers detection procedure. As such, it opens the door to geometric approaches for unsupervised estimations of orientations and camera parameters, making possible fast cryo-EM biomolecule reconstruction.
翻译:冷冻电子显微镜( cryo- EM) 能够生成近原子分辨率的生物分子的重建 3D 图像 。 因此, 它代表了结构生物学中最有希望的成像技术之一 。 然而, 原始的冷冻- EM 图像只是高度腐蚀 - 噪音和带式过滤 - 2D 目标 3D 生物分子显微镜的预测。 重建 3D 分子形状始于清除图像外端, 估计生成给定 2D 图像的生物分子方向, 以及估计相机参数以纠正强度缺陷。 目前, 执行这些任务的技术往往在计算上非常昂贵, 而数据集大小却在不断增长。 需要下一代算法来保持准确性, 同时提高速度和可缩缩缩缩缩。 在本文中, 我们结合变式自动解动的自动立体和感应变式对抗网络( GANs) 来学习低维度潜伏的冷冻- EM 图像的表达方式。 我们对获得的隐性空间进行了探索性分析, 使得这些直隐性空间的图像的定位为“ 感光学” 和感光学的感光学分析结构结构结构 。