论文剖析
生物信息学|DiMeLo-seq:一种用于在基因组范围内绘制蛋白质- DNA相互作用的方法 ///////////////
- 摘要
基因组调控的研究通常使用高通量DNA测序方法来确定特定蛋白质与DNA的相互作用,它们依赖于DNA扩增和短读测序,限制了它们在复杂基因组区域的定量应用。为了解决这些限制,我们开发了长reads测序定向甲基化(DiMeLo-seq),它使用抗体栓系酶在原位将目标蛋白结合位点附近的DNA甲基化。然后,使用长读、单分子测序技术,同时检测这些外源性甲基化标记和未扩增DNA上的内源性CpG甲基化。我们通过绘制人类基因组中染色质结合蛋白和组蛋白修饰的图谱,优化并基准化DiMeLo-seq。。此外,我们确定了着丝粒蛋白A定位在高度重复区域内的位置,这些区域与短测序读数不匹配,我们估计了着丝球蛋白A分子沿单个染色质纤维的密度。DiMeLo-seq是一种通用的方法,为研究蛋白质-DNA相互作用提供多模态、全基因组信息。 2. 介绍
基因组DNA需要由能够读取、调节、复制、重组和修复它的蛋白质进行解码和维护。绘制蛋白质与DNA相互作用的位置以及方式,可以为了解它们在健康和患病细胞中如何发挥功能或故障提供关键的见解。一些强大的方法已经被开发出来来绘制单个靶蛋白与DNA基因组相互作用的位置,包括DamID、染色质免疫沉淀测序(ChIP-seq)。这些方法包括选择性地扩增与特定蛋白质结合的区域的短DNA片段,使用下一代测序(NGS)确定这些DNA分子的序列,并将这些序列映射回参考基因组,使用测序覆盖率作为蛋白质-DNA相互作用频率的测量。虽然这些方法已经被证明对研究DNA结合蛋白和染色质修饰非常有用,但它们也存在一些局限性。
首先,DNA扩增过程不能复制DNA修饰信息,如甲基化和氧化。这就阻止了同时测量蛋白质-DNA之间的相互作用和DNA修饰,并限制了可以收集到的关于这些调节元件之间关系的信息量。其次,基于扩增的富集方法往往依赖于PCR,并具有内在的偏差。因此,这些技术产生的测序覆盖范围仅提供了蛋白质-DNA相互作用频率的半定量读出。
此外,这些方法依赖于消化或剪切DNA成短片段进行富集,然后是NGS,产生短于250碱基对(bp)的测序reads。短片段长度与这些技术通常是必要的,以实现足够的结合位点分辨率。虽然可以在短reads上绘制多个蛋白质-DNA相互作用,但将DNA剪切成短片段会破坏联合的长程结合信息,并阻碍了相位读取来测量单倍型特异性蛋白质-DNA相互作用的能力。此外,人类基因组的重复区域是基因组组装和定位方法的一个主要挑战,因为很难明确地分配短的DNA测序reads到它们在基因组中的独特位置。这些障碍阻碍了我们解决关于重复序列在细胞分裂、蛋白质合成、衰老和基因组调控中的作用的生物学问题的能力。
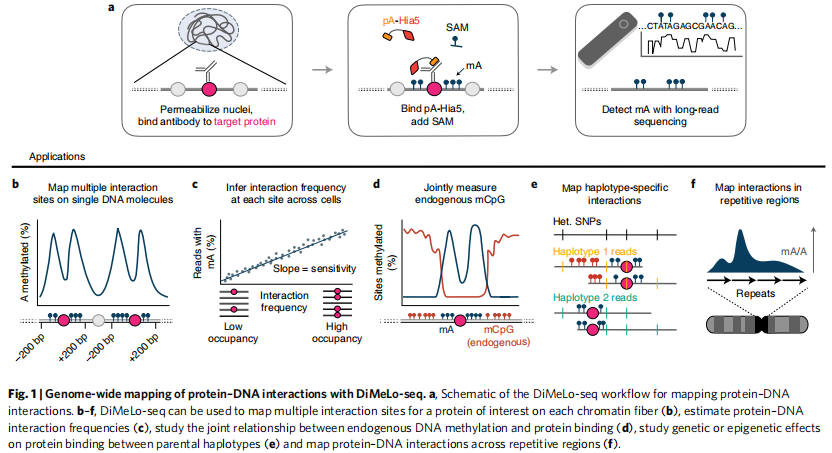
这些局限性突出了对蛋白质-DNA相互作用匹配方法的需求,这些方法必须充分利用长reads单分子测序技术的力量,包括查询组装的重复区域和直接读取DNA修饰的能力。为了满足这一需求,我们开发了DiMeLo-seq。DiMeLo-seq能够在native, long, single, 测序DNA分子上以高分辨率绘制蛋白质- DNA相互作用,同时模拟测量内源性DNA修饰和序列变化。这些特征为以前所未有的方式研究基因组调控提供了机会。最近的技术已经开始利用长reads测序来识别原生单分子上的可访问区域和CpG甲基化,但它们不能直接针对特定的蛋白质-DNA相互作用。在这里,我们扩展了这些功能,通过在包括复杂重复结构域在内的基因组中mapping lamina-associated domains (LADs)、CTCF结合位点、组蛋白修饰/变异和CpG甲基化,来展示DiMeLo-seq的优势。 3. 结果 DiMeLo-seq工作流程。DiMeLo-seq结合了抗体导向的蛋白质-DNA定位方法的元素,将特定目标蛋白附近的甲基化标记沉积起来,然后使用长reads测序直接读出这些外源性甲基化标记。利用人类DNA中N6甲基脱氧腺苷(以下简称mA)的低丰度,我们将抗体结合蛋白A与非特异性脱氧腺苷酸甲基转移酶Hia5(pA–Hia5)融合,以催化靶向染色质相关蛋白附近DNA中mA的形成(图1a)。首先,细胞核被渗透,初级抗体与感兴趣的蛋白质结合,任何未结合的抗体都被洗掉。接下来,pA–Hia5与抗体结合,任何未结合的pA–Hia5都被洗掉。然后将细胞核培养在含有甲基供体S-腺苷蛋氨酸(SAM)的缓冲液中,以激活相关蛋白附近的腺嘌呤甲基化。最后,使用修饰敏感的长读测序法,通过mA碱基调用,提供蛋白质与DNA相互作用位点的读数,对基因组DNA进行分离和测序(图1a和补充图1)。这种方法在检测每个长DNA分子上的多个结合事件方面具有明显的优势,这对于短读测序是不可能的(图1b)。该方案还避免了扩增偏差,从而改进了对细胞群基因组中每个位点的蛋白质-DNA相互作用频率的绝对估计(图1c)。修饰敏感reads可以同时检测外源性抗体在单分子上的腺嘌呤甲基化和内源性CpG甲基化(图1d)。此外,DiMeLo序列的长测序读数经常重叠多个杂合子位点,从而能够对单倍型特异性蛋白质-DNA相互作用进行阶段划分和测量(图1e)。最后,长reads可以绘制基因组高度重复区域内的蛋白质-DNA相互作用(图1f)。
体外重组染色质的抗体导向组蛋白特异性DNA腺嘌呤甲基化。我们表达并纯化重组pA-Hia5,并使用甲基化敏感限制性内切酶DpnI检测其对纯化DNA的甲基化活性,DpnI酶仅在腺嘌呤甲基化时切割GATC位点。在SAM存在的情况下,与Hia5、pA-Hia5或蛋白A/GHia5(pAG-Hia5)孵育的DNA对DpnI消化敏感,证实了纯化的融合蛋白的甲基转移酶活性。为了测试pA-Hia5在体外靶向染色质和甲基化可达DNA的能力,我们使用称为“601”的核小体定位DNA序列重组了含有组蛋白变异着丝粒蛋白A(CENP-A)的染色质。将单核小体与自由浮动pA–Hia5和SAM一起孵化,然后进行长读测序和甲基化敏感碱基调用,显示reads的97.1%±0.8%发生甲基化图2c、d)。此外,我们观察到在预期的核小体保护区域几乎没有甲基化(图2c、d)。
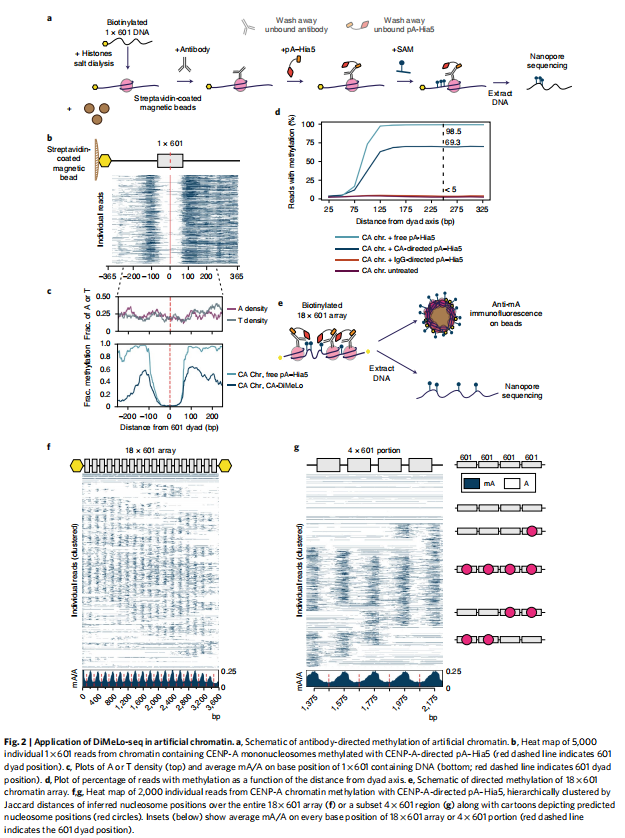
我们在生物素化DNA上重组CENP-A染色质,将其与链霉亲和素涂层的磁珠结合,用CENP-A-抗体和pA-Hia5培养它,并在用SAM激活甲基化之前洗去任何未结合抗体和pA–Hia5(图2a和扩展数据图1c)。我们在65.0%±10.0%的CENP-A DiMeLo-seq reads上观察到甲基化图2b-d),甲基化水平随距离核小体足迹的距离而衰减(图2c)。我们只观察到IgG对照DiMeLo-seq reads的甲基化背景水平(IgG reads的5.1%±0.6%),而未处理的reads数为4.1%±0.5%(图2d)。而从自由漂浮的pA-Hia5或抗体拴系的pA-Hia5条件中读取的数据都显示出核小体大小的甲基化保护(约150-180bp以二分体为中心;图2c,d),从抗体栓系的pA-Hia5上读到的所有甲基化中,约70%发生在二分体两侧的250bp内。这一结果证明了抗体栓系的pA-Hia5可以在体外甲基化靠近靶核小体的可达DNA。
为了测试DiMeLo-seq识别染色质纤维上的目标核小体的特异性,我们首先评估了pA-Hia5甲基化体外重组染色质上的DNA可及区域的能力,该染色质组装在601核小体定位序列的18×阵列上。染色质与自由漂浮的pA-Hia5和SAM共同孵育导致寡核小体足迹的结构模式,正如之前报道的重组染色质与另一种外源甲基转移酶EcoGII孵育。
然后,我们测试了由含有CENP-A或组蛋白H3的核小体重组的染色质阵列的抗体定向甲基化。我们将染色质与CENP-A抗体和pA-Hia5孵育,去除未结合的抗体,并用SAM激活甲基化(图2e)。激活后,我们用抗mA抗体对染色质偶联珠进行免疫染色,显示当CENP-A染色质与pA-hia5和CENP-A抗体孵育时,mA信号增加,表明抗体定向甲基化。长读测序检测到CENP-A定向的CENP-A染色质甲基化后DNA上的mA。平均而言,CENP-A染色质的CENP-A定向甲基化在核小体的中心轴上被耗尽,其中601序列位于核小体二分体的位置(图2f,g)。在个体reads上,我们观察到位于601二分位的甲基化保护,这与核小体占据保护DNA免受抗体定向甲基化一致(图2f,g),与自由pA-Hia5条件相似。与游离pA-Hia5条件下,我们观察到任何不受核小体保护的区域的甲基化发生率很高相比,在抗体导向的pA-Hia5条件下,我们观察到甲基化的平均概率大约低4倍(图2f),这与pA-Hia5的系链产生最接近抗体结合核小体的脱氧腺苷优先甲基化的预期一致。尽管CENP-A DiMeLo序列与游离pA-Hia5处理的序列相比,可访问DNA的总甲基化程度有所降低,但我们在我们的染色质阵列群体中检测到了类似的核小体密度分布。我们使用pAG-Hia5观察到H3抗体定向H3染色质甲基化的类似的结果。我们的结论是,使用组蛋白特异性抗体来指导pA-Hia5的活性是在体外靶向在感兴趣的核小体附近的特异性甲基化。
lamin B1原位映射的优化。接下来,我们优化了DiMeLo-seq,用于在人类细胞系(HEK293T)的每个通透细胞核中原位定位蛋白质- DNA相互作用。为此,我们绘制了lamin B1 (LMNB1)的相互作用位点,LMNB1在DamID研究中经常被用于分析LADs。跨细胞类型的基因组中几乎总是与核层接触的大片区域称为构成层相关结构域(cLADs)。跨细胞类型很少与核膜接触,而是位于核内部的区域称为构成性inter-LAD(ciLAD;图3a)。其他区域在不同细胞类型和/或相同类型细胞之间的lamina接触频率可能不同。我们选择LMNB1作为初始靶点是因为(1)cLADs和ciLADs分别提供了特征良好的靶上和靶外控制区域;(2) LMNB1的结合区非常大(lad中位大小为500kb,大约覆盖30%的基因组),因此即使测序覆盖率很低,也可以检测到DNA-LMNB1的相互作用;(3) LMNB1在核lamina上的定位可以很容易地通过免疫荧光可视化,允许在方案的每个步骤中使用显微镜进行中间质量控制;(4)我们之前已经使用大块和单细胞协议从HEK293T细胞中生成了LMNB1 DamID数据,提供了充足的参考材料。
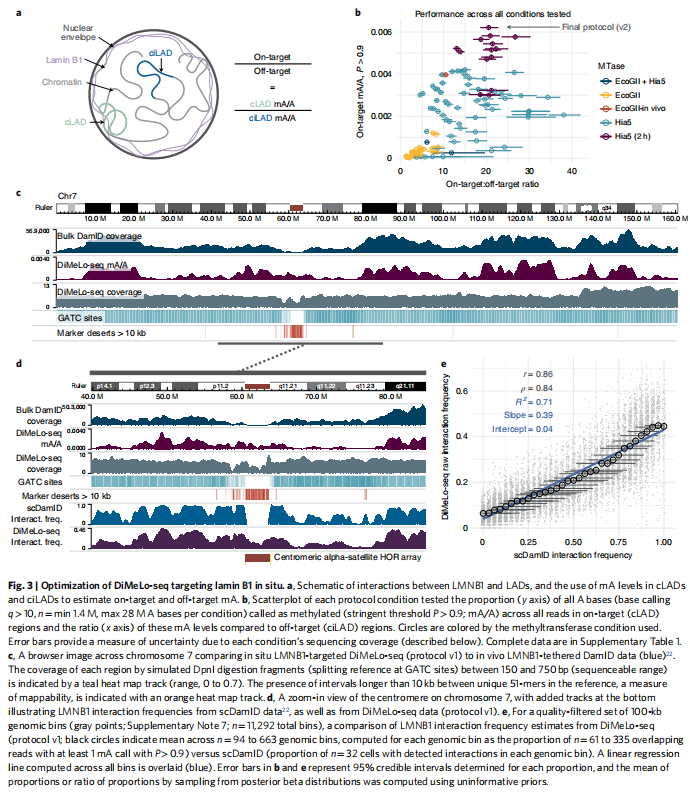
To assess the performance of the LMNB1-targeted DiMeLo-seq protocol, we quantified the proportion of adenines that were called as methylated (mA/A) across all reads mapping to cLADs (on-target regions), and across all reads mapping to ciLADs (off-target regions)。We evaluated the performance of each iteration of the protocol using both the on-target methylation rate (as a proxy for sensitivity) and the on-target:off-target ratio (as a proxy for signal-to-background ratio), aiming to increase both。我们开发了一个快速的管道,用于测试该方案的许多组成部分的变化,允许我们在60小时内从收获的细胞到完全分析的数据。这个优化管道,我们测试了超过100个不同条件(图3b),变化如下:甲基转移酶类型(Hia5和EcoGII),输入细胞数量、洗涤剂、一抗浓度、二抗的使用、酶浓度、孵化温度、甲基化孵化时间,甲基化缓冲液和SAM浓度。我们验证了该方案的初始版本(v1;https://doi.org/10.17504/protocols.io.bv8tn9wn/),,然后进一步优化甲基转移酶激活条件,在不牺牲特异性的情况下增加靶点上甲基化量50-60%(v2;https://doi.org/10.17504/protocols.io.b2u8qezw/)。为了确认这种优化将适用于其他类型的蛋白质,我们还检查了针对蛋白质CTCF的不同协议变异的结果,发现它们是一致的。
We also verified that there is very little loss of performance when using cells that were cryopreserved in dimethylsulfoxide-containing medium or lightly fixed in paraformaldehyde (PFA), when using between 1 and 5 million cells per replicate, or when using concanavalin-A-coated magnetic beads to carry out cell-washing steps by magnetic separation instead of centrifugation。为了确认抗体的特异性,我们分别使用IgG同型对照和自由漂浮的Hia5对照进行了实验,以测量非特异性甲基化和DNA可及性。我们还生成了一个稳定的转导系,在体内表达EcoGII和LMNB1之间的直接融合,如MadID,然后我们通过纳米孔测序检测mAs。与pAG-EcoGII的原位DiMeLo-seq相比,这种体内方法产生了三倍多的靶上甲基化(图3b),尽管这种性能预计会随着不同的融合蛋白及其表达水平而变化。
我们发现,DiMeLo-seq和传统体DamID在基因组的非重复部分高度一致(1-Mb bin中的Spearman相关性=0.71),但传统体DamID在周围熵点区域的覆盖很少甚至没有(图3c)。这在一定程度上是由于周围熵点中可映射短读的唯一序列标记的可用性较低,但也与着心重复中GATC (DamID协议中Dam和DpnI的结合基序)的频率较低有关(图3c)。与DamID不同的是,DiMeLo-seq产生的长读数可以唯一地映射到7号染色体的着丝粒区,揭示了该区域与核薄片有中间水平的接触(图3c,d)。
由于DiMeLo-seq直接探测未扩增的基因组DNA,因此每次测序读数代表单个细胞中的单个自然DNA分子,独立取样,且细胞群体的概率几乎一致。这允许估计绝对蛋白质-DNA相互作用频率,即靶蛋白结合位点的细胞比例,而无需考虑其他蛋白质-DNA映射方法固有的扩增偏差。我们利用来自同一细胞系的单细胞Dam-LMNB1-DamID数据评估DiMeLo序列甲基化与蛋白质-DNA相互作用频率的正交估计之间的关系。这揭示了两个相互作用频率估计值之间的近似线性关系,一个简单的线性模型的R2为0.71,而当将基于单细胞DamID(scDamID)的相互作用频率与大量常规DamID覆盖进行比较时,R2为0.31(图3e)。我们注意到,与DiMeLo-seq相比,scDamID倾向于略微高估中间相互作用频率,这归因于两个协议的体内与原位性质,以及同源物特异性信息在每个低三倍体HEK293T细胞中崩溃的事实。该分析表明,DiMeLo-seq能够估计绝对蛋白质-DNA相互作用频率,而无需考虑扩增偏差,同时在单细胞水平捕获蛋白质-DNA交互作用的异质性。
单分子上CTCF结合和CpG甲基化的联合分析。DiMeLo-seq通过同时检测内源性CpG甲基化、核小体占用和蛋白质结合来测量局部染色质环境下的蛋白质-DNA相互作用。为了突出DiMeLo-seq的这一特征,我们以CTCF为目标,这是一种强烈定位核小体周围的蛋白,其结合被CpG甲基化抑制。我们首先通过计算GM12878 CTCF ChIP-seq峰中甲基化的腺嘌呤相对于这些峰外甲基化的比例,验证了GM12878细胞中CTCF的靶向甲基化是特异性的。我们选择在GM12878细胞中靶向CTCF,因为GM12878是一个编码一级细胞系,具有丰富的ChIP-seq参考数据集。我们测量到,在我们的CTCF靶向样本中,靶向甲基化比背景增加了16倍。我们还测量了CTCF ChIP-seq峰中游离的pA-Hia5对照中6倍的mA/A富集,反映了许多CTCF结合位点与基因组的可访问区域重叠,在这些区域中pA-Hia5更容易甲基化。然而,游离的pA-Hia5和IgG对照组产生的靶向甲基化明显少于CTCF靶向样本。我们通过测量ChIP-seq峰中mA的比例是开放染色质区域,证实了信号富集引起的,而不是CTCF位点的可及性(ATAC-seq)。
为了进一步验证DiMeLo-seq与ChIP-seq数据的一致性,并可视化蛋白质在单个分子上的结合,我们分析了不同强度的ChIP-seq峰内跨越CTCF基序的mA和mCpG(图4a)。DiMeLo-seq信号跟踪与ChIP-seq信号强度,mA密度从ChIP-seq峰值信号的上到下四分位数下降。我们观察到结合基序周围的局部mA增加,从峰中心开始的甲基化周期性衰减,表明强定位的核小体之间相邻连接体DNA的甲基化。结合峰中心88bp的下降反映了CTCF的结合足迹,甚至在单分子上也很明显。通过DNase I足迹和ChIP-exo确定,CTCF与~50bp的DNA结合。DiMeLo-seq观察到的更大的足迹可能是由于Hia5不能有效地在CTCF和DNA物理接触的~20bp内甲基化DNA。我们还观察到一个不对称的甲基化谱,CTCF基序的5‘甲基化更强。这种相对于基序3‘的甲基化增加从中心峰延伸到邻近的连接子DNA。我们推测这种不对称性是由于抗体结合了CTCF的C端而导致的结果,从而将pA-Hia5定位到更接近结合基序的5‘端。为了验证这一假设,我们比较了在使用针对CTCF C端的抗体和针对CTCF N端的抗体时,顶部四分位ChIP-seq峰的结合谱。我们观察到c端结合基序,n端结合基序的甲基化富集(P value=0.00010)。游离的pA-Hia5对照谱支持了这一发现,即抗体结合位点导致了峰的不对称性,因为在这个非靶向病例中没有明显的不对称性。

为了评估DiMeLo-seq对从头峰检测的使用,我们单独使用DiMeLo-seq数据称为CTCF峰,并使用ChIP-seq峰作为真实值,在增加测序深度时创建受试者工作特征曲线。在~25×覆盖率时,我们检测到60%的ChIP-seq峰(假阳性率为1.6%),并测量了曲线下0.92的面积。在DiMeLo-seq检测到的未注释ChIP-seq峰的峰中,10%与hg38参考中1kb标记沙漠和空白重叠,ChIP-seq无法检测到。另外12%的峰位于已知CTCF基序的500bp范围内。
接下来,我们探讨了CTCF结合与内源性CpG甲基化之间的关系。在较弱的ChIP-seq峰中,跨越CTCF结合位点的单个分子在基序周围的mCpG中比在较弱的ChIP-seq峰中表现出较小的下降(图4a)。CpG甲基化和CTCF靶向甲基化之间的反比关系反映了mCpG抑制CTCF结合。我们测量了同一单分子上的mA和mCpG,也观察到A和CpG在连接体DNA中都被优先甲基化(图4b)。连接体DNA中CpG相对于CTCF位点周围的核小体结合DNA的甲基化增加得到了先前研究的支持,这些研究同样报道了连接体DNA中的mCpG水平高于CTCF位点周围的核小体DNA。
CTCF已知的结合基序和丰富的全基因组使其成为表征DiMeLo-seq分辨率的良好靶点。为了表征分辨率,我们估计了跨越CTCF ChIP–seq峰顶部十分位的单分子上的峰中心。平均单分子峰中心为CTCF基序中心的6bp 5′,约70%读数的峰中心在基序中心±200bp范围内。这种预测基序峰中心5′的系统性偏差可以用靶向CTCF C末端时观察到的甲基化不对称性来解释。影响DiMeLo-seq分辨率的另一个因素是甲基转移酶的范围,可以通过测量峰中心甲基化密度的衰减率来表征。为此,我们将关于模体中心的平均腺嘌呤甲基化密度拟合成指数函数,并计算出169bp的半衰期。总的来说,该分析表明DiMeLo-seq可以将结合事件解析到大约200bp以内;然而,这一指标可能依赖于蛋白质靶点,并受局部染色质环境的影响。
为了描述DiMeLo-seq检测单分子上CTCF结合事件的敏感性,我们根据CTCF峰区内每个reads的甲基化腺嘌呤的比例,对个体CTCF靶向的DiMeLo-seq读取进行了二分类,定义为CTCF结合基序中心周围的±150bp。对于前十分位的ChIP-seq峰,这是最有可能包含CTCF结合的区域,我们对包含CTCF结合事件的reads进行了分类,敏感性为54%(5.7%的假阳性率)。
接下来,我们研究了DiMeLo-seq测量单分子上相邻位点的蛋白质结合的能力。我们首先描述了CTCF在单个分子所跨越的两个结合位点上的占用情况。我们能够检测到在两个位点或仅在两个位点之一上被CTCF结合的邻近CTCF基序,并且检测到的结合似乎与ChIP-seq峰强度有关(图4c)。这一分析证明了DiMeLo-seq在分析长单分子上的配位结合模式方面的潜力,而这对于短reads方法是不可能实现的。我们进一步研究了6号染色体上一个特定HLA位点的这种潜力,在该位点中,CTCF结合基序中的单倍型特异性单核苷酸多态性(SNPs)阻止了CTCF在两个相邻位点之一的结合。DiMeLo-seq可以绘制单倍型特异性的相互作用,因为长reads通常跨越多个杂合子位点,允许reads被分阶段进行。重要的是,在25×的覆盖范围内,我们能够检测到同一单一分子上的两个位点的结合模式,并可以将在两个位点中的一个检测到的结合缺失归因于结合基序内的突变。绘制单倍型特异性相互作用的能力也有助于研究印迹基因组区域,如IGF2/H19印迹控制区域,在该区域,父等位基因上的CpG甲基化阻止CTCF结合,而在母等位基因上,CTCF能够结合(图4d)。我们还报道了特定位点的单倍型特异性CTCF结合谱,并广泛横跨活性和非活性X染色体。这些结果表明,DiMeLo-seq可以测量单倍型特异性遗传或表观遗传变异对蛋白质结合的影响。
为了测试DiMeLo-seq与其他能够修改调用的长reads测序平台的兼容性,我们对CTCF靶向的DiMeLo-seq样本和未甲基化对照的DNA进行了PacBio测序。我们使用两种方法发现了相似的富集谱,表明DiMeLo-seq与PacBio的循环一致测序技术兼容。然而,虽然PacBio测序报告提高了碱基呼叫的准确性,但这种方法在未甲基化对照中检测到更多的甲基化,略微降低了测量的信噪比。
定位着丝粒区域的蛋白质-DNA相互作用。用DiMeLo-seq定位异染色质中的组蛋白修饰。为了测试DiMeLo-seq在基因组异色重复区域测量蛋白质占用的能力,我们以三甲基化组蛋白H3赖氨酸9(H3K9me3)为目标,它在中心周异染色质中丰富。我们选择在HG002细胞中靶向H3K9me3,因为这个雄性来源的淋巴母细胞系9的X染色体着丝粒已经完全组装好,而且有许多不同的测序数据类型。为了验证靶向甲基化的特异性,我们计算了HG002 CUT&RUN H3K9me3峰中甲基化腺嘌呤的比例,与广义峰外甲基化腺嘌呤的比例。对于HG002细胞中的H3K9me3靶向,mA/A在CUT&RUN峰中的富集是背景的3.6倍(图5a),表明基因组预期的含H3K9me3区域的甲基化富集。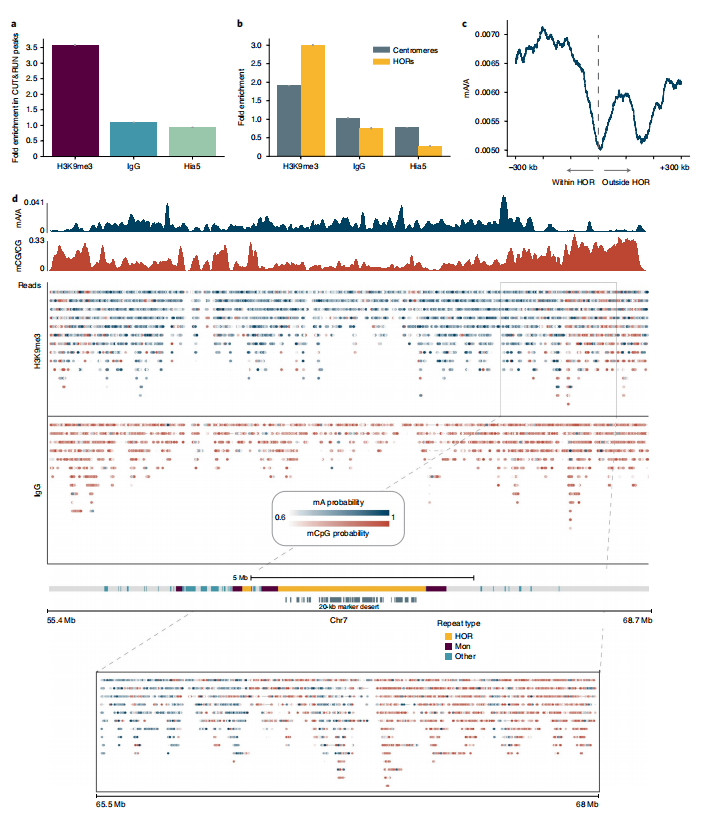

人类的着丝粒位于高度重复的阿尔法卫星序列中,这些序列被组织成高阶重复序列(HORs)。为了验证h3k9me3定向的mA信号在着丝粒中的富集,特别是在HOR阵列中,我们同样计算了mA/A的增加倍,发现在非着丝粒区域富集1.9倍于着丝粒,在活性(着丝粒结合)HOR阵列富集3倍(图5b)。接下来,我们观察了HOR阵列的边界,并观察到H3K9me3从HOR阵列内部到外部移动的边界的下降(图5c)。相比之下,对于游离的pA-hia5对照,随着染色质变得更容易获得,mA/A从HOR阵列内外的移动而增加(扩展数据图9a)。
我们不仅在HOR阵列边界上绘制了异染色质,而且在跨着丝粒的单分子上。以h3k9me3为靶点的DiMeLo-seq读取图谱横跨7号染色体的着丝粒,甚至在独特标记之间超过20kb的区域(图5d)。一个IgG同型对照证实,h3k9me3靶向样本中的腺嘌呤甲基化不是由背景甲基化引起的(图5d)。与依赖于扩增短DNA片段的方法不同,如ChIP-seq和CUT&RUN,我们能够检测染色质边界的单分子异质性,如从65.5到68Mb的过渡,随着CpG甲基化的增加,H3K9me3信号下降(图5d)。然而,异染色质的甲基化效率较低,以及在重复区域绘制中等长的reads的挑战,仍然会导致这些区域的不均匀和低覆盖率。为了提高靶向DiMeLo-seq在着丝粒中应用的敏感性,我们开发了一种着丝粒富集方法来增强活性HOR阵列的覆盖率,并将该方法应用于CENP-A的研究。
基于限制的富集策略提高着丝粒覆盖率。在alpha卫星HOR阵列中,着丝粒特异性组蛋白变体CENP-A描绘了功能着丝粒和动粒形成的位置。群体水平研究表明,CENP-A-核小体位于这些重复单元的核心,其中重复是最同源的。然而,目前还不可能通过解析CENP-A核小体在单个染色质纤维上的位置来确定CENP-A在着丝粒的一维结构和密度。为了使用DiMeLo序列绘制CENP-A核小体在着丝粒的位置,我们制定了一种策略,专门为人类着丝粒DNA进行富集,以避免对整个基因组进行测序。
我们的富集策略称为AlphaHOR RES,它基于经典的着丝粒富集策略,包括使用限制酶消化基因组,限制酶经常切割着丝粒区域外但很少切割着丝束区域内,然后去除短DNA片段。我们将AlphaHOR RES添加到DiMeLo seq工作流中,观察到着丝粒处的测序覆盖率至少增加了20倍,同时保持了相对较长的读取长度(平均值~8kb;图6a、b)。因此,这种富集策略大大增加了测序的分子比例,有助于研究CENP-A分布,节省了大量测序时间和成本。此外,因为AlphaHOR RES针对的是DNA而不是蛋白质与DNA的相互作用,并且因为它是在定向甲基化完成后进行的,所以不太可能偏离我们对这些区域中蛋白质与DNA相互作用频率的推断。

DiMeLo-seq显示了跨着丝粒间可变的CENP-A核小体密度。我们对HG002细胞进行了CENP-a定向的DiMeLo-seq实验。提取总基因组DNA后,我们在测序前使用AlphaHOR-RES对着丝粒序列进行富集(图6a,b)。以一种与比对无关的方式,我们根据一个可用的短读测序数据集中的CENP-a富集的k-mers的存在或缺失,对DiMeLo-seq序列进行了分类。与不含CENP-A的k-mers相比,富含CENP-A的k-mers的CENP me-seq的腺嘌呤甲基化程度大约增加了7倍(图6c)。当比较CENP-A靶向样本与游离的pA-Hia5样本时,我们观察到含有CENP-A k-mers的DiMeLo-seq reads中类似的绝对甲基化水平。然而,游离的pA-Hia5样品在不含CENP-A k-mers的reads中也有更高的mA/A比例,这表明在缺乏靶向性的情况下缺乏CENP-A特异性。
为了检测CENP-A核小体在着丝粒重复阵列中的位置,我们将我们的reads与一个完整的人类杂交组装体对齐,其中包含一个来自HG002细胞系的完整组装的染色体X。我们研究了最近描述的染色体X着丝粒倾斜区(CDR),这是着丝粒alpha HOR阵列中的一个低甲基化区域,其中短读CENP-A数据集对齐。我们证实了CDR内源性CpG甲基化(图6d)。与相邻的CpG甲基化区域相比,CENP-A导向的mA值都更高,这与该细胞系的短读数据一致(图6e,f)。我们发现,与邻近区域相比,在X染色体CDRs内检测到的CENP-A核小体的密度增加了5倍(图6g)。我们估计,26%的±5%的核小体在染色体X CDR中含有CENP-A,而只有5%的±2%的核小体在邻近区域内含有CENP-A图6g)证实了集成短读方法不能做到的情况:CDRs中单个DNA分子上的CENP-A核小体的密度增加。IgG同型对照证实,该信号不是由于背景甲基化(2%±1%)。在染色体X CDR内的IgG对照reads上检测到的核小体(图6g)。先前的一项研究估计,内源性人类着丝粒的平均CENP-A密度为25个核小体中的1/1,假设平均着丝粒大小为~1Mb45。相比之下,我们估计,在x染色体上较小的~100kb的CDR中,至少有1/4的核小体含有CENP-A。这表明CENP-A核小体的占用在人类着丝粒中差异很大。此外,我们还发现CENP-A密度最高的区域与CDR相一致。我们在3号染色体上观察到类似的CENP-a定向甲基化分布,其中两个HOR阵列中只有一个有明显的CENP-a定向甲基化。这些观察结果支持了在每条染色体上发现一个活跃的HOR阵列。这些发现阐明了在单个染色质纤维上的HOR序列中CENP-A核小体的密度和位置,这是以前现有技术无法实现的。 4. 讨论 在这里,我们开发、优化和验证了DiMeLo-seq,一种用于绘制全基因组范围内蛋白质-DNA相互作用的长reads方法。DiMeLo-seq可以在已测序的DNA的单分子上的数百个碱基对上定位蛋白质的结合位点,长度高达数百个碱基点。这种长读取提高了基因组高度重复区域的可定位性,为未来对其调控和功能的研究打开了大门。由于DiMeLo-seq不涉及扩增,它可以用来更好地估计基因组中每个位点的蛋白质-DNA相互作用的绝对频率。它还提供了关于内源性CpG甲基化和在同一长单分子上的蛋白质-DNA相互作用的联合信息,这些信息可以分阶段揭示单倍型特异性的结合和甲基化模式。
通过将单个CENP-A核小体定位到长的、已测序的DNA分子上,我们发现在着丝粒内mcpg缺失区域的单个染色质纤维上的CENP-A核小体密度增加。CENP-A DiMeLo-seq体外CENP-A染色质的敏感性测量~65%,表明估计CENP-A核小体密度在染色体X CDR下限,和实际CENP-A密度在CDR甚至可能高于~25%(图6g)。CENP-A位置变化的一个来源是染色质的细胞周期状态。因为先前存在的CENP-A核小体被认为在每个细胞周期中通过表观遗传学指导新的CENP-A核小体的组装,所以了解在细胞周期同步后,CENP-A密度如何沿着活性着丝粒的序列变化将是一件有趣的事情。
我们估计DiMeLo-seq对CTCF和LMNB1的单分子敏感性在54%到59%之间,与非靶区相比,阈值达到94%的特异性。然而,灵敏度可能因目标蛋白和抗体而异,这可能是由于局部空间效应的差异,或目标蛋白、抗体或pA结合强度的差异。
这项研究还使我们能够描述使用DiMeLo序列与短读集成方法相比的优势和权衡。因为DiMeLo-seq是一种无扩增的方法,可以对单个天然DNA分子进行测序,并且因为它依赖于离心来进行清洗步骤,所以它需要相对大量的起始材料来产生足够大的细胞颗粒,以便于处理(每次复制100至200万个细胞)。使用我们证明与DiMeLo-seq协议兼容的刀豆球蛋白-A涂层磁珠,可能有助于未来减少这些细胞输入要求。此外,标准的DiMeLo-seq协议要求对整个基因组进行统一测序,这可能会浪费与目标蛋白结合域无关的基因组区域的测序读数。对于仅靶向小区域的蛋白质,可以进行靶向DNA测序或使用DNA富集方法,如AlphaHOR RES,这里展示的着丝粒富集方法。另一组最近描述了一种互补的方法,使用一组不同的限制性内切酶来富集着丝粒DNA,这可能是AlphaHOR-RES的重要替代品。也可以使用免疫沉淀来富集甲基化DNA或与感兴趣的蛋白质结合的DNA,但这将不再从细胞群中均匀地采集DNA分子,潜在地削弱了从测量的甲基化频率推断蛋白质-DNA相互作用频率的能力。
因为Hia5倾向于甲基化未结合的连接子DNA,DiMeLo-seq提供了关于局部核小体占据以及目标蛋白的足迹的信息。这也意味着高度不可接近的区域可能更难以甲基化,而且它们可能需要更高的测序覆盖率。此外,由于DiMeLo-seq是在保持染色质构象的条件下原位进行的,如果它在三维(3D)空间中足够接近目标蛋白的结合位点,它可能会反式甲基化,CUT&RUN也是如此。这些3D相互作用,以及介导它们的因素,可以在进行DiMeLo-seq之前通过扰乱3D染色质结构来研究,这也可能是提高高度浓缩区域DNA可及性的有用方法。
我们预计DiMeLo-seq将有助于研究广泛的生物学问题。例如,因为它可以让一个探索蛋白质的密度沿着一个染色质纤维从一个细胞,它可以用来研究染色质状态之间的边界不同单细胞,或者DNA结合蛋白的化学计量学增强剂影响附近基因的转录。我们还证明了DiMeLo-seq可以读出通过体内表达蛋白和MTases融合沉积的甲基腺烯,如传统的DamID或MadID,而不是原位靶向抗体。这可能被证明有助于研究更多短暂的蛋白质-DNA相互作用,或缺乏合适抗体的蛋白质,如果正在研究的生物系统可以很容易地进行基因修饰。我们也可以想象添加外源性胞嘧啶甲基化标记,以提供关于DNA可及性或第二种蛋白质结合谱的联合信息。虽然我们在本研究中主要使用了牛津纳米孔技术测序,但我们也证明了DiMeLo-seq与PacBio高保真(HiFi)测序兼容,这可能是需要高度准确的碱基调用的首选,如基因组组装。通过这项研究,我们发现DiMeLo-seq提供了一种通用的方法来表征跨越难以查询的相互作用的单个分子。 原论文名称:DiMeLo-seq: DiMeLo-seq: a long-read, single-molecule method for mapping protein–DNA interactions genome wide