
编译 | 王海云
审稿 | 王娜 本文介绍由清华大学生命科学学院生物信息学教育部重点实验室、北京结构生物学高级创新中心和生物结构前沿研究中心、合成与系统生物学研究中心的Qiangfeng Cliff Zhang通讯发表在 Nature Communications 的研究成果:作者提出了SCALEX,一种深度学习方法,通过将细胞投射到一个批次不变的、共同的细胞嵌入空间,以真正的在线方式(即不需要重新训练模型)整合单细胞数据。SCALEX在不同模式的基准单细胞数据集(scRNA-seq,scATAC-seq)上的表现大大优于在线iNMF和其他最先进的非在线整合方法,特别是对于有部分重叠的数据集,在保留真正的生物差异的同时准确地对齐类似细胞群。作者通过构建人类、小鼠和COVID-19患者的可持续扩展的单细胞图谱来展示SCALEX的优势,每个图谱都由不同的数据源组装而成,并随着每个新数据的出现而不断增长。在线数据整合能力和卓越的性能使SCALEX特别适合于大规模的单细胞应用。

简介
单细胞实验可以将样本分解为不同的细胞类型和细胞状态。许多计算工具已经被开发出来用于单细胞数据集的综合分析,所有这些工具都是为了从非生物噪音中分离出生物变异,如不同供体、条件和/或分析平台的批次效应。
目前大多数的单细胞数据整合方法都是基于跨批次的细胞对应关系的搜索,例如类似的单个细胞或细胞群。这些方法有三个限制。首先,它们容易混合只存在于某些批次的细胞群,这对于整合每批中包含不重叠的细胞群的复杂数据集(即部分重叠的数据)是一个严重的问题。其次,它们需要的计算资源随着细胞数量和批次的增加而急剧增加,使得这些方法越来越不适合今天的大规模单细胞数据集。最后,这些方法只能消除当前正在评估的数据集的批次效应。每次增加一个新的数据集,都需要一个全新的整合过程,改变以前研究的现有整合结果。在线数据整合能力在当今的单细胞实验中变得越来越关键。
在这里,作者开发了SCALEX,作为一种基于VAE框架的异质单细胞数据的在线整合方法。SCALEX的编码器被设计成一个数据投影函数,在投影单细胞时只保留批量不变的生物数据成分。重要的是,该投影函数是一个通用的函数,不需要对新数据进行再训练,因此允许SCALEX以在线方式整合单细胞数据。
结果
SCALEX实现了一个通用的编码器,能够在线整合单细胞数据 为了实现在线整合,SCALEX的基本设计理念是实现一个广义的投影函数,将单细胞数据的批处理相关成分从批处理不变量成分中分离出来,并将批处理不变量成分投影到一个共同的细胞嵌入空间。在这里,为了获得一个无需重新训练的用于数据投影的通用编码器,SCALEX包括三个具体的设计元素(图1a)。首先,SCALEX实现了一个没有批处理的编码器,它只从输入的单细胞数据(x)中提取与生物相关的潜在特征(z),以及一个特定批处理的解码器,它通过在数据重构期间将批处理信息纳入其中,从z中重构原始数据。只向解码器提供批次信息,使编码器只关注学习批次不变的生物成分,这对编码器的泛化能力至关重要。第二,SCALEX在其解码器中包括一个使用多分支批次归一化的DSBN层,以支持在单细胞数据重建过程中纳入批次特定变化。第三,SCALEX编码器采用了一个迷你批次策略,从所有批次(而不是单一批次)中取样,这更严格地遵循了输入数据的整体分布。请注意,每个迷你批次都要经过编码器中的批次归一化层,以调整每个迷你批次的偏差,并使其与整体输入分布相一致。
SCALEX比最先进的单细胞数据整合方法要准确得多 作者按照最近一项比较研究中提出的评价框架,广泛评估了SCALEX的基本数据整合性能。作者在多个数据集上与多种方法做了比较,包括在线iNMF和一些最先进的非在线单细胞数据整合方法,包括Seurat v3、Harmony、MNN、Conos、BBKNN、Scanorama、LIGER(即批量iNMF)和scVI。作者根据基准数据集,通过统一模态逼近和投影(UMAP)嵌入可视化以及一系列评分指标,评估了这些工具的整合性能。
MNN、scVI和Conos整合了许多数据集,但仍有一些常见的细胞类型没有得到很好的排列。在线的iNMF、LIGER、BBKNN和Scanorama经常有未合并的共同细胞类型,有时还错误地将不同的细胞类型混在一起。例如,考虑到PMBC数据集中两批之间的T细胞群(图1b),虽然SCALEX、Seurat v3、Harmony、MNN、scVI整合是有效的,但在线iNMF将一些CD4初始T细胞与CD8初始T细胞错位,并将一些NK细胞与CD8 T细胞错位。根据调整兰德指数(ARI)和归一化互信息(NMI)的评估,SCALEX在细胞类型聚类方面的表现大大优于其他所有方法(图1c)。
SCALEX可扩展到Atlas级别的数据集,并可容纳 不同的数据模式 在最近的一项比较研究中,包含大量细胞并由来自多个组织的异质和复杂样本组成的单细胞数据集被称为 " Atlas-level "数据集。这些数据集对数据整合工具提出了新的挑战。作者将SCALEX应用于一个典型的Atlas数据集,即人类胎儿Atlas数据集,其中包含来自GSE156793和GSE134355两个数据批次的4,317,246个细胞,以此来测试SCALEX的可扩展性和计算效率。SCALEX准确地整合了这两批数据,显示了相同细胞类型的良好排列(图1d)。SCALEX可用于整合其他模式的单细胞数据(如scATAC-seq、通过测序对转录组和表位进行细胞索引、CITE-Seq等)和跨模式的数据(如同时分析scRNA-seq和scATAC-seq)。SCALEX在整合小鼠大脑scATAC-seq数据集方面大大优于所有其他方法(图1f),并在整合其他单细胞数据模式包括CITEseq和空间转录组MERFISH数据方面表现良好。作者还用SCALEX整合了一个跨模式的数据集(scRNA-seq和scATAC-seq),发现SCALEX正确地整合了两种模式的数据,并区分了scRNA-seq数据中特有的稀有细胞,包括pDC和血小板细胞(图1g),根据UMAP嵌入和多种分析指标,SCALEX的表现比其他方法更好,包括scjoint和bindSC。
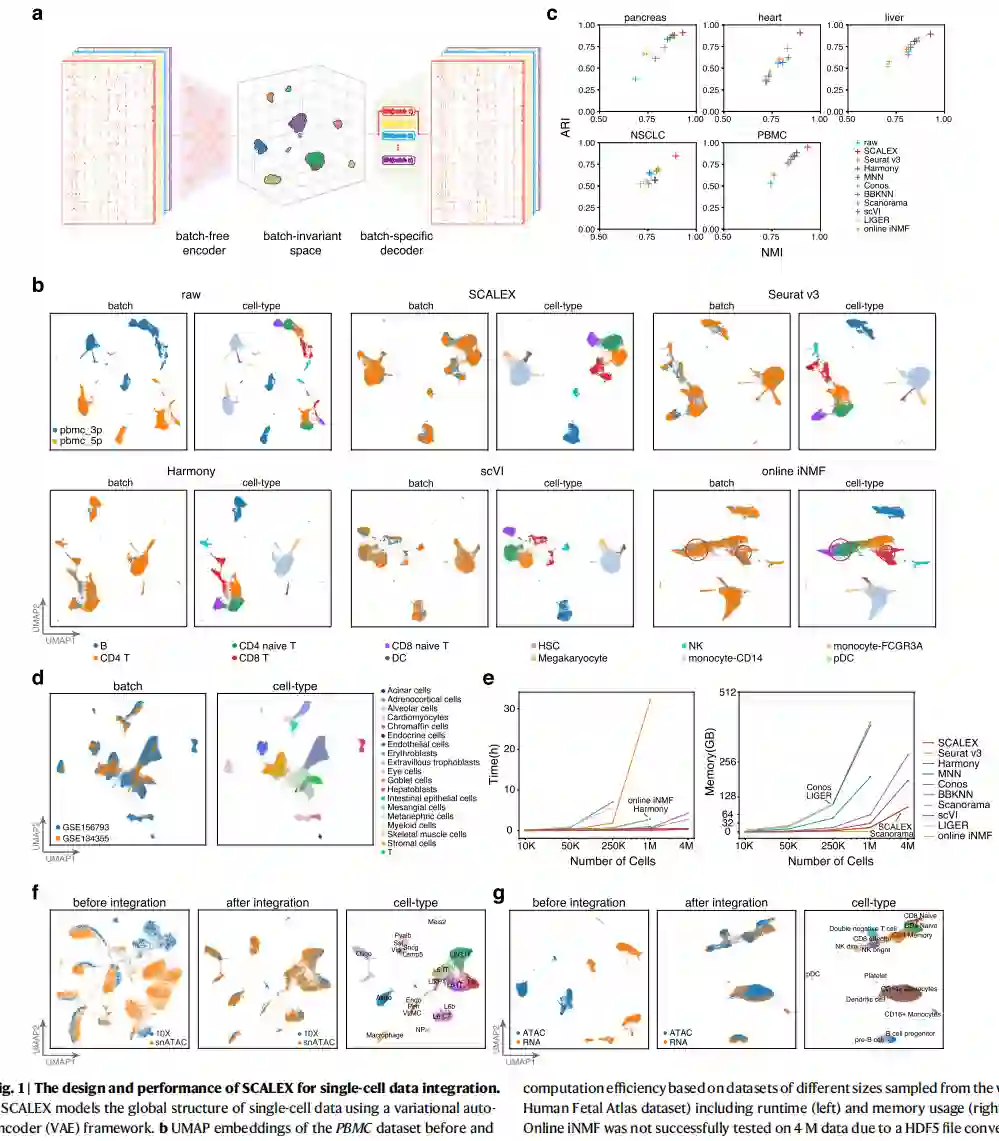
图1 用于单细胞数据整合的SCALEX的设计和性能。
SCALEX在没有过度修正的条件下整合了部分重叠的数据集 许多最近的单细胞数据集,特别是Atlas级数据集,具有高度的样本异质性和复杂的细胞类型组成。这些数据集往往包含部分重叠的批次,其中每批包含一些不重叠的细胞群。这种部分重叠问题给单细胞数据整合带来了重大挑战,往往会导致过度校正的问题(即不同细胞类型的混合),特别是对于那些基于局部细胞相似性的方法。例如,Seurat v3混合了肝细胞CXCL1、肝细胞-CYP2A13和肝细胞TAT-AS1细胞,Harmony混合了肝细胞-CYP2A13和肝细胞-TAT-AS1细胞(图2a)。作为一种将细胞投射到一个共同的细胞嵌入空间的全局整合方法,预计SCALEX对这个问题不那么敏感。事实上, SCALEX正确地保持了五个肝细胞亚型的分离(和scVI一样,图2a)。出乎意料的是,尽管是一个全局性的方法,在线iNMF严重遭受了过度修正,混合了所有五个肝细胞亚型,甚至混合了B细胞和NK细胞(图2a),可能是因为其矩阵因子算法强迫不同细胞类型的对齐。
作者定义了一个过度修正分数,这是一个衡量这种过度修正问题的指标,基于每个细胞的邻域中细胞类型不一致的百分比。从形式上看,过度校正得分是一个负指数,即过度校正得分越高,细胞类型的不准确混合程度越严重。对于基准数据集,SCALEX的过度校正分数最低(图2b),而在线iNMF产生了极高的过度校正分数。为了系统地描述不同方法在部分重叠的数据集上的性能,作者构建了具有一系列常见细胞类型的测试数据集,这些数据集是根据胰腺数据集中六种主要细胞类型的下采样产生的。SCALEX整合在所有情况下都是准确的,对准相同的细胞类型而没有过度校正,而Seurat v3、Harmony和在线的 iNMF经常混合不同的细胞类型(图2c,d)。
SCALEX通过在线投影添加新的数据,增加了现有细胞空间的范围和分辨率 SCALEX的编码器具有通用性,可以将不同来源的细胞投射到一个共同的细胞嵌入空间,而不需要重新训练模型,这使得SCALEX能够以在线方式将新的单细胞数据与现有的数据进行整合。作者对SCALEX基于胰腺数据集的新到数据的在线数据整合性能进行了测试。在投影之前,作者首先使用SCALEX来整合胰腺数据集,这准确地消除了原始数据中明显的批次效应(图3a)。随后,作者使用在原始胰腺数据集上训练的同一个SCALEX编码器将三批新的胰腺组织scRNA-seq数据(图3b)投影到这个 "胰腺细胞空间"。投影后,新批次中的大多数细胞都准确地对准了胰腺细胞空间中的正确细胞类型,从而能够通过细胞类型标签转移对其进行准确注释(图3c)。作者通过计算ARI、NMI和F1分数来评估投影的准确性,以便通过标签转移与原始研究中的细胞类型信息进行细胞类型注释。作者将结果与在线iNMF和scVI进行了比较,这两个工具能够将细胞投影到现有的细胞空间中(注意scVI的数据投影需要通过scArches进行模型再训练)。在与在线iNMF和scVI的比较中,SCALEX取得了最高的投影精度(图3c)。scVI也取得了较高的精度,将大多数细胞投影到正确的位置,只有少数α和导管细胞例外。在线iNMF在纳入新的批次时混合了不同的细胞类型,例如将一些α细胞投射到γ和δ细胞的位置上,这反过来又导致了在标签转移时的错误注释(图3c)。
将新的单细胞数据投射到现有的细胞嵌入空间的能力,使SCALEX能够随时用额外的信息细节来丰富(即增加生物分辨率)这个细胞空间。为了验证这一点,作者将另外两批黑色素瘤数据(SKCM_GSE72056,SKCM_GSE123139)投射到先前构建的PBMC空间。同样,SCALEX正确地将所有常见的细胞类型投射到PBMC细胞空间的相同位置(图3d),但在线iNMF将肿瘤细胞与血浆、单核细胞和CD8 T细胞混合,scVI则将CD8T细胞分成几个不同的组。重要的是,对于只存在于黑色素瘤数据批次中的肿瘤细胞和浆细胞,SCALEX没有将这些细胞投射到PBMC空间中的任何现有细胞群上;相反,它将它们投射到靠近类似细胞的新位置,浆细胞投射到靠近B细胞的位置,而肿瘤细胞投射到靠近造血干细胞的位置(图3e)。这表明SCALEX可以通过数据投影,用新的细胞类型充实现有的细胞空间 通过数据投射来丰富现有的细胞空间。
SCALEX投影还可以利用新数据对现有细胞空间中的未知细胞类型进行事后注释。例如,作者注意到在胰腺数据集中有一组以前未被描述的细胞(图3a)。作者发现这些细胞显示了已知上皮基因标记物的高表达水平。因此,作者从支气管上皮细胞数据集中收集了一些上皮细胞,然后将这些上皮细胞投射到胰腺细胞空间。作者发现,一组抗原呈递的气道上皮细胞(SLC16A7+上皮细胞)被投射到未定性细胞的同一位置(图3f)。这些数据,再加上观察到这两个细胞群显示出类似的标记基因表达(图3g),表明这些未定性细胞也是SLC16A7 +上皮细胞。
SCALEX整合构建可扩展的单细胞图谱 将异质数据结合到一个共同的细胞嵌入空间的能力使SCALEX成为一个强大的工具,从不同的数据集集合中构建一个单细胞图集。尽管原始数据有很强的批次效应,SCALEX还是准确地将三批小鼠图谱的数据整合到一个共同的细胞嵌入空间中(图4a-c)。常见的细胞类型在细胞空间的同一位置排列整齐,包括所有组织中的B、T和内皮细胞,以及特定组织中的近端肾小管、尿路细胞和肝细胞。不同的细胞类型被单独定位,如Microwell-seq数据中的精子、Leydig和小肠细胞,Smart-seq2数据中的角质细胞干细胞和大肠细胞,表明生物变化被很好地保存下来。作者将SCALEX与其他所有方法进行了比较,发现SCALEX在细胞类型聚类方面表现最好,尤其是避免了过度校正(图4d,e)。重要的是,用SCALEX生成的图谱可以通过投影新的单细胞数据来进一步扩展,以支持原始图谱和新数据中的细胞的比较研究。为了说明这一效用,作者将来自Tabula Muris Senis(Smart-seq2和10X)的两批额外的老年小鼠组织数据和两个单组织数据集(肺和肾)投射到SCALEX小鼠图谱的细胞空间。作者发现,新的数据批中的细胞被正确地投射到初始图集的细胞包埋空间中相同的细胞类型的位置上(图4f),这一点被标签转移对新数据的准确细胞类型注释所证实(图4g)。
一个综合的SCALEX COVID-19 PBMC图谱揭示了COVID-19患者之间不同的免疫反应 许多单细胞研究已经被用于分析COVID-19患者的免疫反应。然而,这些研究往往存在样本量小和/或对各种疾病状态采样有限的问题。为了进行全面的研究,作者使用SCALEX生成了一个COVID-19 PBMC图谱,整合了9项COVID-19研究的数据,涉及10批共860,746个单细胞(图5a)。作者确定了22种细胞类型,每种类型都有基因表达数据支持的典型标志物(图5b,c)。有趣的是,作者发现一些细胞亚群与病人状态有不同的关联(图5d)。CD14单核细胞亚群(CD14-ISG15-Mono)的特点是高表达I型干扰素刺激的基因(ISG)和富含免疫反应相关基因本体论(GO)术语的基因(图5e,f)。从轻度、中度到重度患者,CD14-ISG15 Mono细胞的频率明显增加(图5g)。在COVID-19患者中,作者观察到在轻度、中度和重度病例之间,CD14-ISG15-Mono细胞的ISG基因表达明显下降,表明在重度COVID-19患者中存在类似免疫衰竭的反应(图5e)。
SCALEX COVID-19 PBMC Atlas与SC4联盟研究的在线整合 作者基于SCALEX COVID-19 PBMC图谱的分析结果与中国COVID-19单细胞联盟(SC4)研究的两个结论一致,该研究最近进行了大规模的努力,从171名COVID-19患者和25名健康对照者中产生了一个超过100万个细胞的单细胞图谱。首先,这两项研究观察到相同的免疫细胞亚群,显示出与COVID-19严重程度的不同关联。CD14单核细胞、巨核细胞、浆细胞和原T细胞的比例随着疾病严重程度的增加而升高,而pDC和mDC细胞的比例下降(图5g)。其次,根据在SCALEX COVID-19 PBMC Atlas中的细胞计算相同的细胞因子评分和炎症评分(在SC4研究中定义),作者证实单核细胞亚群与SARS-Cov2感染引发的细胞因子风暴有关,并且在严重患者中进一步升高(图5j)。
SCALEX的在线整合能力使作者能够将SC4联盟的数据集投射到SCALEX COVID-19 PBMC图谱的细胞空间。作者发现,两个图集的细胞类型排列得很好(图5h,i)。SC4数据的整合进一步大幅提高了SCALEX COVID-19 PBMC图谱的范围和分辨率。首先,该数据将巨噬细胞和上皮细胞加入到细胞空间中,使调查它们在COVID-19中的潜在参与成为可能。这种整合还支持对特定的细胞亚群进行更精确的表征。例如,巨核细胞群在SCALEX COVID-19 PBMC图谱或SC4图谱中都没有区分,在SC4投影后,在联合图谱中被分为两个亚群(图5h)。对这两个新划分的巨核细胞亚群(TUBA8-Mega和IGKC-Mega)中差异表达的基因进行探索性功能分析,发现IGKC-Mega细胞的GO术语 "体液免疫反应 "富集,但TUBA8-Mega细胞的 "血小板激活的负性调节 "富集(图5k)。这些结果说明了使用SCALEX生成的可持续扩展的单细胞图谱如何利用现有的大规模数据资源,并促进新的生物和生物医学见解的发现。
3 总结与讨论 单细胞研究正变得越来越普遍,规模越来越大,样本类型的范围也在不断扩大,往往有相当多的异质细胞子集。因此,非常需要数据整合工具来准确和有效地处理这些Atlas级的数据集。此外,还需要有在线整合能力,以不断地将传入的新数据与现有的整合相结合,而不必从头开始重新计算。SCALEX学习了一个广义的投影函数,将异质的单细胞数据投影到一个共同的细胞嵌入空间,使其能够实现真正的在线数据整合。SCALEX在计算上也是高效的,并且在整合部分重叠的数据集时保留了生物变化,避免了过度校正。这些特点使SCALEX对Atlas级别的数据集特别有用,允许整合许多单细胞研究,以支持整个生命科学和生物医学领域正在进行的、非常大规模的研究项目。作者推测,使用SCALEX来预测来自高度多样化的癌症类型的单细胞数据集,以构建一个泛癌症单细胞图谱,可能会导致发现以前未知的细胞类型,这些细胞类型在不同的癌症中是共同的,并在发病机制、恶性肿瘤进展和/或转移中发挥作用。
参考资料 Xiong, L., Tian, K., Li, Y. et al. Online single-cell data integration through projecting heterogeneous datasets into a common cell-embedding space. Nature Communications 13, 6118 (2022). https://doi.org/10.1038/s41467-022-33758-z
代码 https://github.com/jsxlei/SCALEX
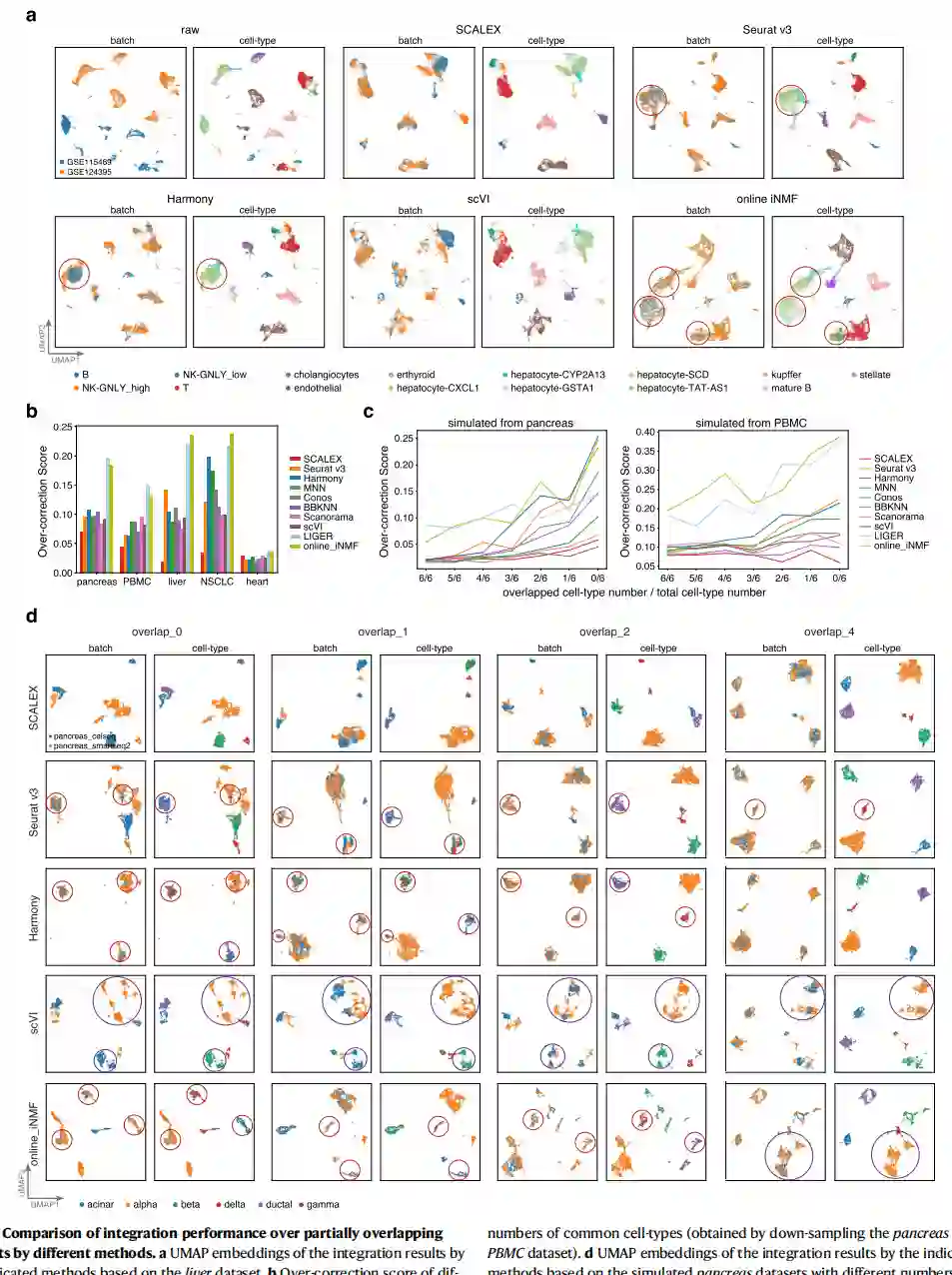
图2 不同方法对部分重叠的数据集的整合性能比较。
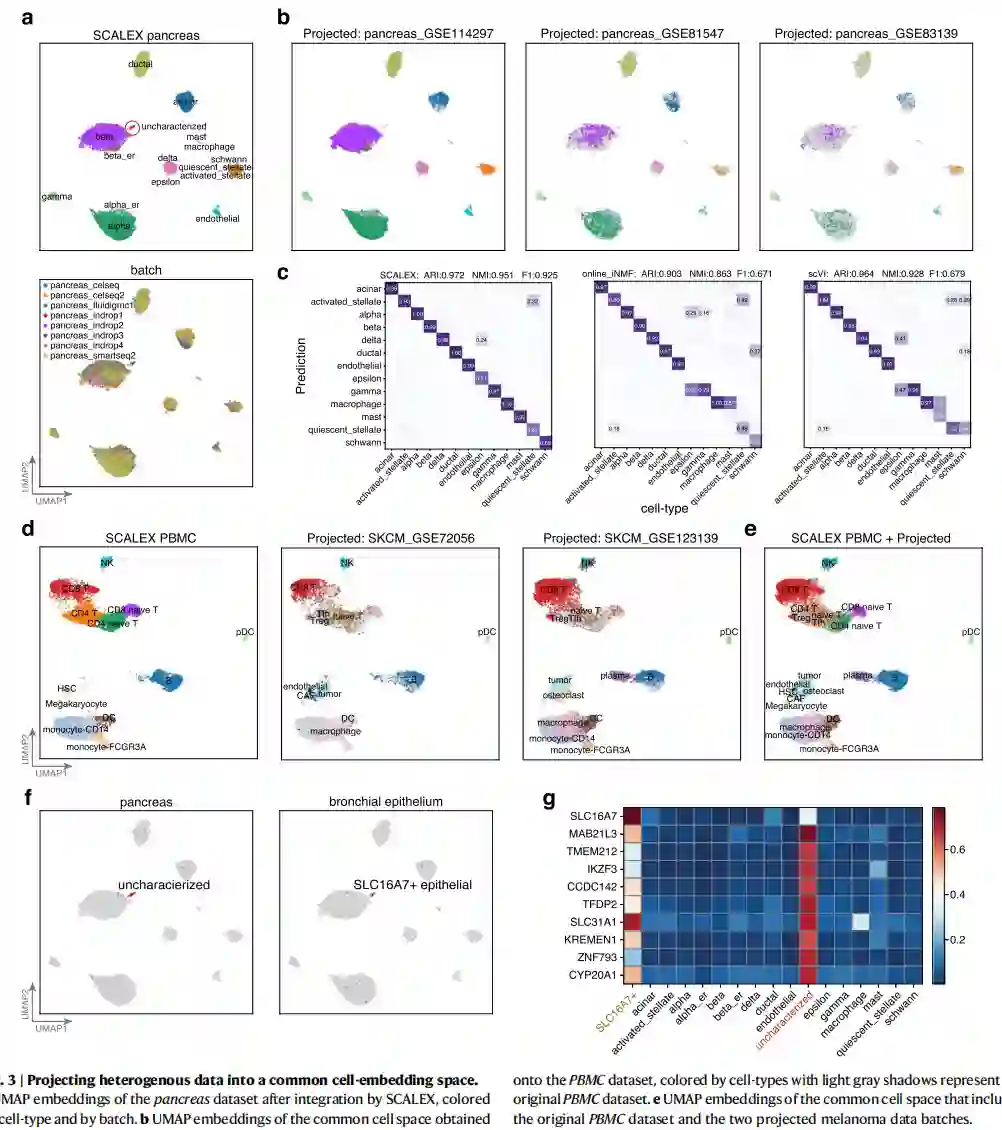
图3 将异质数据投射到一个共同的细胞嵌入空间中。
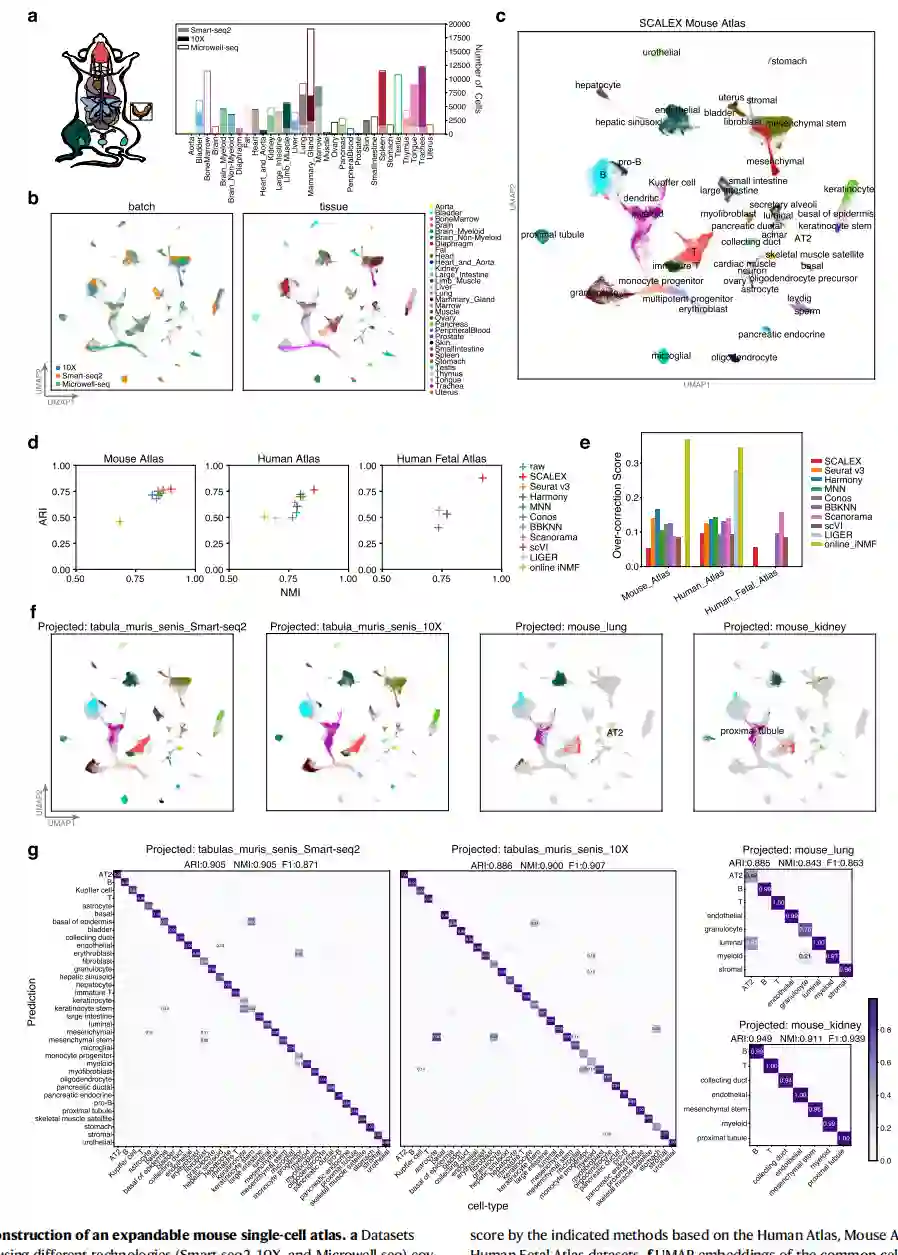
图4 构建一个可扩展的小鼠单细胞图谱。
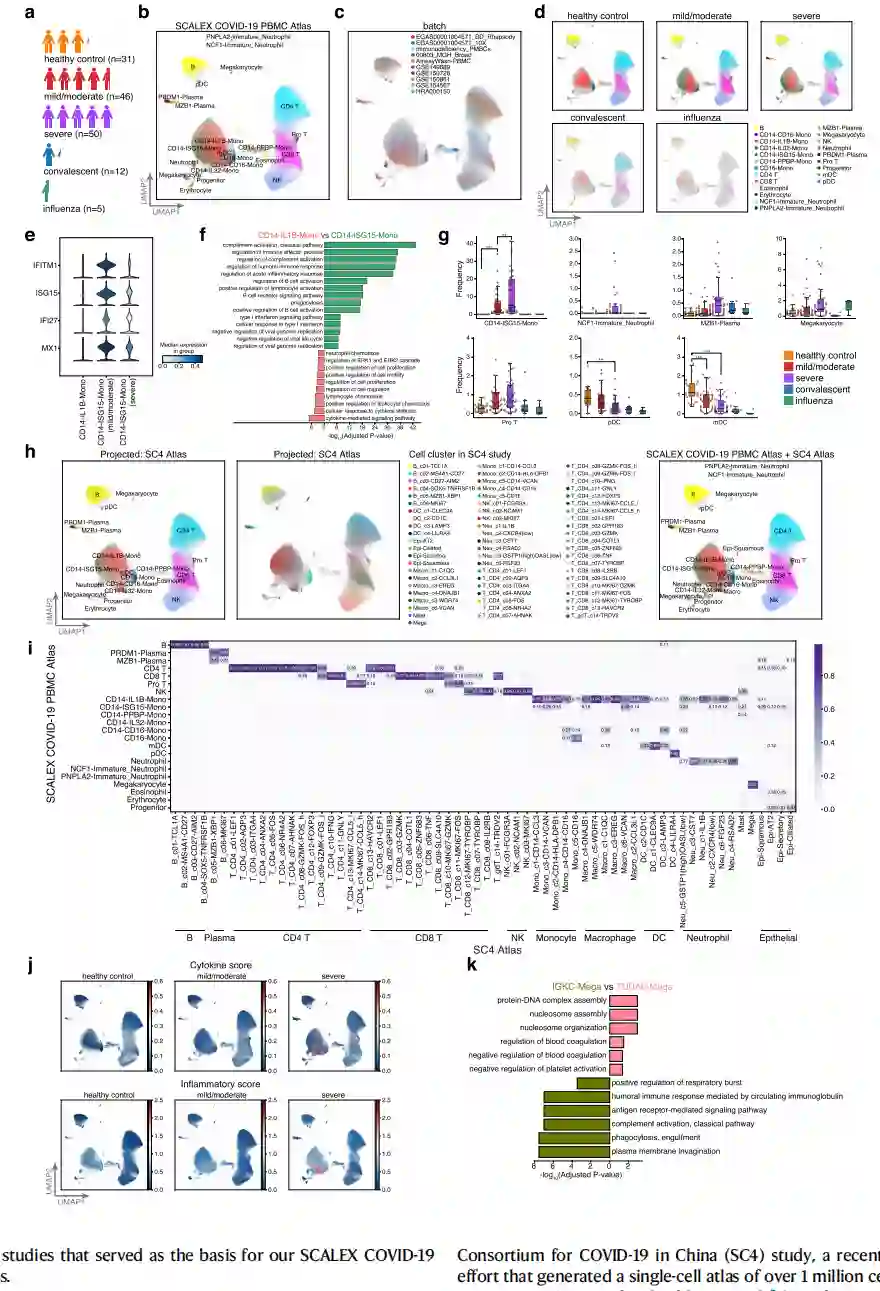
图5 在线整合COVID-19 PBMC图谱。
