
2022年5月30日,来自韩国首尔国立大学生物科学学院的Martin Steinegger和哈佛大学FAS科学部的Sergey Ovchinnikov等人在Nat Methods杂志发表文章,介绍了一个快速和易于使用的蛋白质结构预测工具ColabFold。 ColabFold通过将MMseqs2的快速同源搜索与AlphaFold2或RoseTTAFold相结合,提供了蛋白质结构和复合物的加速预测。ColabFold的搜索速度提高了40-60倍,并且优化了模型的利用,在一台有图形处理单元的服务器上每天可以预测近1000个结构。与Google Colaboratory相结合,ColabFold成为一个免费的、可获得的蛋白质折叠平台。

ColabFold开源:https://github.com/sokrypton/ColabFold 新型环境数据库:https://colabfold.mmseqs.com ColabFold 为了利用AlphaFold2和RoseTTAFold这些方法的力量,研究人员需要强大的计算能力。 首先,为了建立多样化的多序列比对 (MSA),需要使用最敏感的同源检测方法HMMer和HHblits搜索来自公共参考和环境数据库的大量蛋白质序列。这些环境数据库包含了从元基因组和转录组实验中提取的数十亿的蛋白质。由于其庞大的规模,搜索一个单一的蛋白质可能需要几个小时,且需要超过2TB的存储空间。其次,为了执行深度神经网络,即使是相对常见的约1000个残基的蛋白质大小,也需要具有大量GPU RAM (随机存取存储器) 的GPU。然而,对于这些,MSA的生成主导了整个运行时间。 为了使没有这些资源的研究人员能够使用AlphaFold2,研究人员开发了基于谷歌Colaboratory的独立解决方案。Colaboratory是一个由谷歌托管的Jupyter笔记本的专有版本。登录的用户可以免费使用它,并且可以使用强大的GPU。同时,Tunyasuvunakool等人为Google Colaboratory开发了一个AlphaFold2 Jupyter笔记本 (称为AlphaFold-Colab),其输入的MSA是通过用HMMer搜索UniProt Reference Clusters (UniRef90) 和一个8倍还原的环境数据库而建立的。这导致预测的准确性较低,同时仍然需要很长的搜索时间。 本文提出了ColabFold,一个快速和易于使用的软件,用于预测蛋白质结构和同质和异质复合物,可作为Google Colaboratory内的Jupyter笔记本使用,在研究人员的本地计算机上作为笔记本或通过命令行界面使用。ColabFold通过用快40-60倍的MMseqs2 (多对多序列搜索) 取代AlphaFold2的同源性搜索,加快了单个预测的速度,并通过避免重新编译和增加早期停止标准,使批量预测的速度提高了约90倍。结果表明ColabFold在CASP14目标上优于AlphaFold-Colab并与AlphaFold2相匹配,在ClusPro数据集上的预测质量也与AlphaFold-multimer相匹配。 ColabFold的组成 ColabFold (图1) 由三部分组成。第一部分是一个基于MMseqs2的同源搜索服务器,用于建立不同的MSA和寻找模板。该服务器有效地将输入序列与UniRef100、PDB70和环境序列集进行比对。第二部分是一个Python库,它与MMseqs2搜索服务器进行通信,为结构推断 (单链或复合物) 准备输入特征,并使结果可视化。这个库还实现了一个命令行接口。最后一部分由使用Python库的Jupyter笔记本组成,用于基础、高级和批量使用。
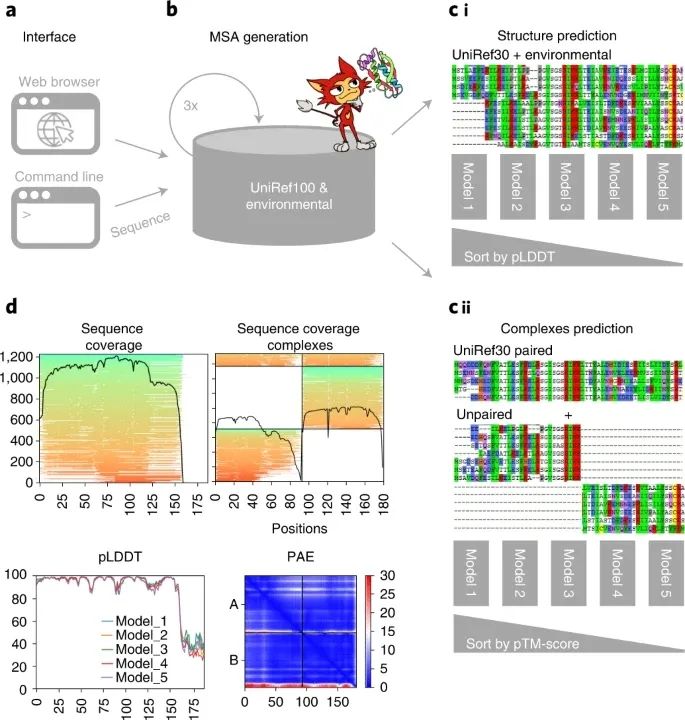
图1:ColabFold的示意图
几点改进 ColabFold用MMseqs2取代了敏感的搜索方法HMMer和HHblits。ColabFold优化了MMseqs2的MSA生成,使其具有以下三个特性:MSA的生成应该是快速的、MSA必须很好地捕捉多样性、它必须小到可以在内存有限的计算机上运行。虽然第一个要求是通过快速的MMseqs2预过滤器实现的,但对于第二和第三个要求,研究人员开发了一个搜索工作流程,以最大限度地提高灵敏度和一个新的过滤器,对序列空间进行均匀采样。预测质量取决于输入的MSA,然而,通常一个MSA只有几个 (约30个) 足够多样化的序列就足以产生高质量的预测。 此外,研究人员将HHblits和HMMer在AlphaFold2中分别使用的Big Fantastic Database (BFD) 和MGnify数据库合并为一个冗余度降低的版本,称之为BFD/MGnify。鉴于真核生物蛋白质的多样性在BFD和MGnify数据库中没有得到很好的体现,环境搜索数据库提供了一个改善非细菌序列结构预测的机会。由于复杂的内含子和外显子结构,组装和基因调用的局限性导致参考数据库中的代表性不足。因此,研究人员对BFD/MGnify进行了扩展,增加了包含真核生物蛋白的元基因组蛋白目录、噬菌体目录和MetaClust的更新版本。这个数据库被称为ColabFoldDB。结果显示,与BFD/MGnify相比,ColabFoldDB对蛋白质家族数据库 (Pfam) 中小于30个成员的结构域产生更多样化的MSA。 预测准确性比较 为了比较预测结构的准确性,研究人员比较了AlphaFold2 (带模板的默认设置)、AlphaFold-Colab (无模板)、ColabFold-RoseTTAFold-BFD/MGnify、ColabFold-AlphaFold2-BFD/MGnify和ColabFold-AlphaFold2-ColabFoldDB对CASP14竞赛中所有目标的模板建模分数 (TM-分数) (图2a)。所有三种ColabFold模式都是在没有模板的情况下执行的。在考虑到MSA生成 (图2b) 和模型推理的情况下,ColabFold的单次预测平均比AlphaFold2和AlphaFold-Colab快5倍。
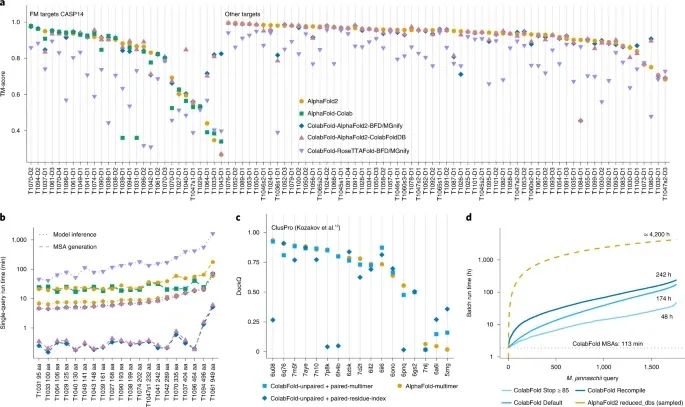
图2:单链和复合物的预测比较 ColabFold-AlphaFold2-BFD/MGnify、ColabFold-AlphaFold2-ColabFoldDB、AlphaFold2、AlphaFold-Colab和ColabFold-RoseTTAFold-BFD/MGnify的自由建模目标的平均TM分值分别为0.826、0.818、0.79、0.744和0.62。在所有的CASP14目标上 (不包括AlphaFold-Colab,因为它不能作为一个独立的方法),各方法的TM分值分别为0.887、0.886、0.888和0.754。如果MMseqs2的成分过滤器被禁用,ColabFold对目标T1084的预测可以从TM-分数0.457提高到0.872。
AlphaFold2最初发布时没有建立蛋白质复合物模型的能力。然而,研究人员发现,通过将两个序列与甘氨酸连接体结合起来,它常常可以成功地建立复合物模型。 对于高质量的预测,有研究表明序列应该以成对的形式提供给AlphaFold2。研究人员实施了一个类似的配对程序,并在图2c中显示了ColabFold对复合物的预测能力。ColabFold在ClusPro数据集上用AlphaFold-多聚体模式预测复合物时达到了最高的准确性,然而,有些目标用残基指数模式表现更好。 对于高通量结构预测,研究人员在ColabFold中引入了几个功能。首先,MSA的生成可以在批处理模式下独立于模型批处理推断执行。第二,只编译五个AlphaFold2模型中的一个,并重复使用权重。第三,避免对相似长度的序列进行重新编译。第四,实施早期停止标准,以避免额外的循环或模型,如果一个足够准确的结构已经被发现。最后,开发了命令行工具colabfold_batch来预测本地机器上的结构。 总之,结果表明,短于1,000个氨基酸的1,762个蛋白质组成的Methanocaldococcus jannaschii蛋白质组可以在48小时内完成预测,在一台Nvidia Titan RTX上的pLDDT (预测的局部距离差异测试;每个残基的置信度) ≥85时可以提前停止 (图2d),同时几乎没有牺牲预测的准确性。在50个蛋白质的子样本中,AlphaFold2和ColabFold Stop≥85的平均pLDDTs为89.75和88.78。 ColabFold在AlphaFold2的基础上,改进了序列搜索,提供了同源和异源复合物的建模工具,扩展了高级功能,扩大了环境数据库,并实现了大规模批量预测蛋白质结构,速度比AlphaFold2提高了约90倍。 参考资料 Mirdita, M., Schütze, K., Moriwaki, Y. et al. ColabFold: making protein folding accessible to all. Nat Methods (2022). https://doi.org/10.1038/s41592-022-01488-1
--------- End ---------