
作者 | 杨千立 审稿 | 陈梓豪 指导 | 闵小平(厦门大学) 此次为大家分享的是来自Nature Communiations 上的一篇题为”Predicting the structure of large protein complexes using AlphaFold and Monte Carlo tree search” 的文章,来自斯德哥尔摩大学生物化学和生物物理系的Patrick Bryant团队。
AlphaFold可以以非常高的精度预测单链和多链蛋白质的结构。然而随着链数的减少,预测准确性降低,GPU显存限制了可以预测的蛋白质复合物大小。作者团队表明可以从子组件的预测开始预测大型复合物的结构。使用蒙特卡洛树搜索从预测的175个子组件中收集了91个复合物,其具有10-30个链,其TM-score中间值为0.51。其中有30个高精确的复合物 (TM-score ≥ 0.8)。作者团队还创建了一个评分函数mpDockQ,该函数可以区分装配是否完整并预测其准确性。结论发现包含对称性的复合物被精确组装,而不对称复合物仍然具有挑战性。
1 介绍 蛋白质复合物支配许多细胞过程,例如mRNA剪接,蛋白质降解或辅助蛋白质折叠。通过结合来自许多共纯化实验的蛋白质相互作用信息,人类蛋白质复合物图谱hu.MAP 2.0提供了4779个具有多于两条链的复合物。然而,PDB中仅存在83种这些复合物。只有372个具有超过两条链的人类蛋白质复合物进行了结构解析,并且在3130个真核生物核心复合物中,只有800个具有覆盖PDB中所有链的同源结构,这表明作者对蛋白质复合物的结构知识存在差距。
至少有三种方法对蛋白质复合物的结构进行建模。即使已知每种单体的结合形式,使用AlphaFold进行二聚配合物的折叠和对接方法也优于基于模板的建模和对接方法。此外,很少有对接程序处理多于两条链,这些方法不适合构建与已知复合物没有密切同源性的大型复合物。目前没有用于多于两条链的复合物的对接基准,并且以前的研究仅报告了几个示例的结果。用集成模型组装大型蛋白质复合物通常需要电子密度图或其他实验信息来指导组装过程,然而,由于某些蛋白质复合物难以表达,纯化和结晶,因此很难获得电子密度。
唯一用于预测多于两条链的蛋白质结构的深度学习方法是AlphaFold-multimer。该方法已经在多达九条链或具有1536个残基的蛋白质上进行了训练,并且可以预测多达几千个残基的复合物,其中GPU显存限制起作用。然而,对于具有多于两条链的蛋白质,性能迅速下降。因此,预测较大配合物的结构目前不可行。另一种方法可能是预测大型复合物的子组件的结构,然后组装它们。作者早些时候已经表明,在少数情况下可以从二聚体中手动组装大型复合物。
在体内,大型蛋白质复合物的所有部分都不会同时组装,而是逐步组装,这是由于存在同源蛋白质链和潜在的界面,需要在添加后续链之前将其掩埋。
在这里,作者探讨了AlphaFold预测具有10-30条链的蛋白质复合物的局限性,并创建了一种图遍历算法,该算法排除了重叠的相互作用,从而可以逐步组装大型蛋白质复合物。
2 结果 下面介绍使用蒙特卡洛树搜索 (MCTS) 的组装蛋白质复合物组装的流程。作者使用AlphaFold-multimer 2.0 (AFM)或基于AlphaFold的FoldDock协议以探索成功率。
2.1复合物装配(组装路径) 为了分析组装大型蛋白质复合物的可能性,作者从PDB中提取了总共175个高分辨率的非冗余复合物,其具有多于九条链,这些链不包含来自不同生物体的核酸或相互作用。作者首先分析组装这些蛋白质复合物的可能性,假设蛋白质链之间的确切配对相互作用是已知的。使用AFM或FoldDock,作者预测所有作为蛋白质子组件的相互作用蛋白质链对,并从中创建组装路径(Assembly path)。
作为示例,6ESQ (乙酰乙酰-CoA硫解酶/HMG-CoA合酶复合物) 的组装在图1中示出,使用AFM预测的子组件。从两个二聚体C-A和H-C开始,使用两个二聚体中共同存在的链C通过叠加产生三聚体H-C-A。接下来,通过与H的连接来添加链L (使用链H的叠加); 之后,通过与L的连接来添加链J; 此过程继续进行,直到根据轮廓路径组装整个复合体。
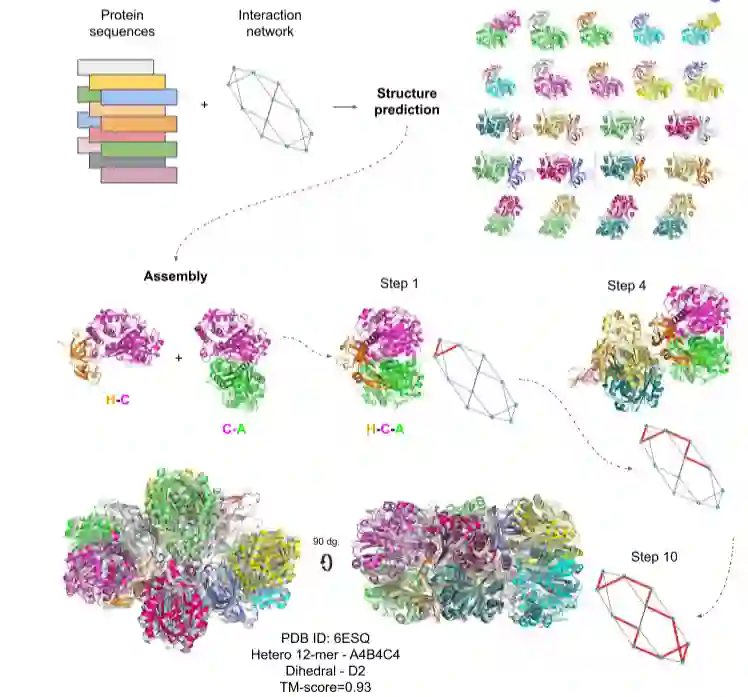
图1.复合物6ESQ的组装规则
所有相互作用链的结构由来自每条链的蛋白质序列和相互作用网络预测。这些预测作为指导来构造装配路径。在每个步骤中,通过网络边缘添加一条新链,从而顺序地构建了复杂的结构。路径用红色标注。装配的复合物在原生复合物上用灰色展示出来。使用来自AFM的子组件得到0.93的TM-Score(如图),使用FoldDock得到0.92的TM-Score(未展示)。
在本文中,作者假设交互图是已知的,即作者将组装路径限制为仅包括复合物中存在的交互。尽管经过这样简化,组装路径的数量仍然很大,并且在理论上可以从其他类型的实验或预测中获得此信息。接下来,通过从随机选择的链开始并通过叠加所有可能的连接来创建潜在的组装路径。由于不完善的子组件,预测之间经常发生重叠,从而导致例如来自链B和C的原子在给定的A-B-C-D复合物中中占据相同的空间位置。因此,当来自两个不同链的超过一半的 α 碳在彼此之间5å以内时,组装路径就会中断。当复合体中的所有链都可以链接在一起时,组装路径就完成了。
2.2蒙特卡洛树搜索 由于要探索的可能组装路径数量众多,搜索所有路径都是不可行的。因此,作者使用MCTS搜索最优路径,从随机选择的链 (节点) 开始,随机添加链以扩展路径,从而创建新的节点。从这些扩展中,模拟了完整的装配。当无法添加其他子组件时,将停止拓展。模拟程序通过mpDockQ进行评分,并将分数反向传播以产生对先前选择的支持。选择支持最多的路径,创建一个最有可能正确的复合物。由于搜索过程的统计性质,在反向传播中没有 “学习” 特定复合物的任何方面,即所有175个复合物都可以用于评估。
图2.MCTS用于搜索组装路径
3.3 使用成对交互中AFM与AF的比较 175个复合物中只有15个可以利用AFM和FoldDock两者的天然成对相互作用完成组装。基于FoldDock和AFM的组件是部分互补的,这意味着AFM 或者FD的子组件可用于组装复合物。结果表明,如果可以找到完整的路径,则与FoldDock模型 (TM中位数 = 0.77) 相比,AFM的模型略好 (TM中位数 = 0.83)。
AlphaFold-multimer (AFM-v1) 建模流水线经常引起冲突,导致来自不同链的原子占据相同位置,这就是为什么开发新版本 (AFM-v2) 。将冲突定义为来自彼此在1 å 内的不同蛋白质链的原子,AFM二聚体具有26.7% (175/656)冲突 ,FoldDock pipeline具有6.3% (41/656)冲突。AFM预测中存在更多冲突,但最终组件的质量也更高,这表明子组件是准确预测的,有关详细信息,请参见原文。
3.4 二聚体中的有限构象采样 在组装过程中,由于预测不完全正确,不同蛋白质链的加性相对方向可能导致重叠。组装过程中重叠的一个原因是由于并非正确预测链相互作用的所有构象,从而导致某些二聚体中的界面错误或缺失。例如,作者研究了1A8R,在预测独特的成对相互作用时,只能找到一种类型的二聚构象,但是在复合物中,每条链与其他链至少具有两种不同类型的相互作用。这意味着不可能从预测的二聚体组装整个复合物。这里,可以通过预测三聚体相互作用来规避重叠界面,从而生成替代界面。因此,作者继续预测三聚体亚组,并使用三聚体中的所有二聚体进行组装。
3.5 使用三聚体相互作用的复杂装配 使用FoldDock协议和AFM可以预测所有复合物的所有天然三聚体相互作用。使用天然三聚体,冲突比使用二聚体更频繁,AFM产生37.1% (829/2234)冲突,而对于FoldDock pipeline,产生23.2% (520/2242)冲突。从三聚体中提取所有二聚体相互作用,并像前文一样构建组装路径。在175个复合物中,有58 (33%) 和55 (31%)个复合物可以组装完成,其中FoldDock和AFM的TM分数中位数分别为0.80和0.74 (图3a)。与引导的二聚体TM评分相比,引导的三聚体方法产生了46个额外的复合物,并且FoldDock的TM评分中位数更高。对于AFM,通过引导三聚体方法产生了43个额外的复合物,缺少了3个额外的复合物,但中值TM得分低于二聚体。
在许多情况下,不知道所有蛋白质链的确切相互作用,仅知道一组链的相互作用。在将MCTS应用于作者假设了相互作用知识的蛋白质复合物之后,作者现在转向在不知道相互作用的情况下预测复合物。除了可能错误地识别出交互对的问题之外,这还大大增加了可能的错误路径的数量。无论如何,作者发现91/175 (52%)的复合物可以以0.51 (图3a) 的中位数TM分数组装,而对于AFM,只有74个复合物以TM中位数为0.61完成。
当两种三聚体方法应用FoldDock进行装配,完全和天然三聚体方法的分数中位数分别为0.76和0.80(53个复合体)。对于AFM,相应的分数是0.77和0.79 (45个复合体)。因此,使用本机交互会导致总体得分略高,但完整程序集的分数较低。FoldDock生成的模型总体上优于AFM,这主要是因为AFM的完整装配较少 (图3a)。作者还与Multi-LZerD和Haddock进行了比较,提供了真实的链结构作为输入。对于Haddock,以TM分数中位数0.29完成77个复合物)。对于Multi-LZerD,作者无法完成数据集中任何复杂的对接。
为了分析当一个复合物组装完成并具有高TM分数 (≥ 0.8,n = 30) 时的可能性,作者分析ROC曲线 (图3b) 作为的函数;,创建mpDockQ分数,结果表明mpDockQ趋于高时,复合体的TM分数和完整性也是如此 (图3c)。这表明mpDockQ可用于选择复杂的完成时间以及精确程度。
图3d显示了使用FoldDock组装复合物的TM分数分布以及在不同TM分数阈值下的示例。配合物的对称性类型和分组的准确性强烈地影响了结果。发现具有对称性的配合物可以进行高精度地组装,显示了MCTS对对称配合物的适用性。获得具有非常高TM分数 (≥ 0.8) 的完整复合物是至关重要的,因为不完全正确的大型复合物不太可能提供生物学上有意义的见解。还可能的是,一些具有低TM分数的复合物是准确的组装,但其构象与PDB中的构象不同 (例如5TRM具有八面体对称性,但以二面体构型组装)。
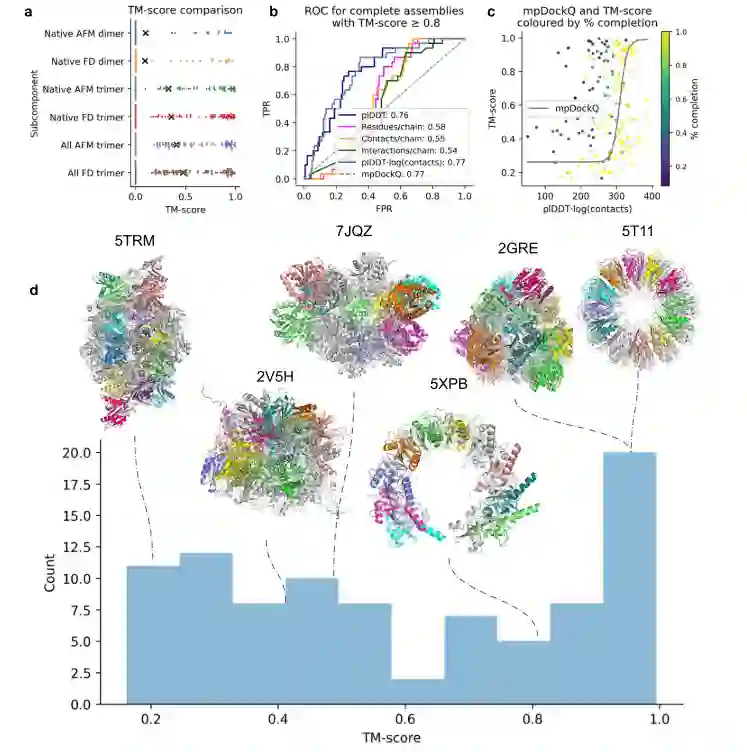
图3.使用多种方法的复杂复合物组装
3.6 专配具有4~9条链的复合物 MCTS的组装方法也可能用于较小的复合物。为了分析提高AFM端到端 (E2E) 在较小复合物上的准确性的可能性,作者创建了一组具有4-9条链的278个复合物。作者团队使用FoldDock预测了所有可能的三聚体子组件,并使用MCTS组装了它们。作者发现AFM E2E的性能在4-9链的所有低聚物中都高于MCTS组装 (图4)。但平均而言,该性能较低,这表明仍有很大的改进空间,即使预测很小的蛋白质复合物的结构仍尚未解决。
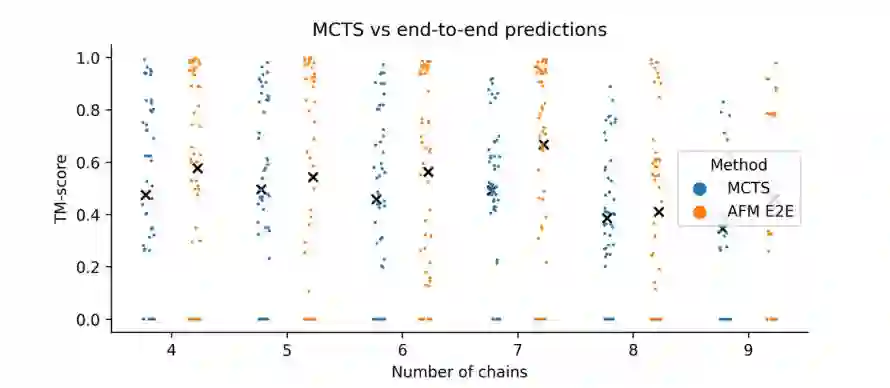
图4.MCTS与AFM E2E在小复合物上的比较
3 总结与讨论 直接从序列信息预测大型复合物的结构目前是一项艰巨的挑战。本文中提出了一种方法,预测子组件并将其组装成更大的复合体。使用从天然二聚体、天然三聚体和所有可能的三聚体相互作用预测的子组件,TM得分中位数分别为0.77、0.80和0.50。评分函数mpDockQ可以区分装配是否完整并预测其准确性。基于AF的FoldDock在预测三聚体亚组分方面优于AFM,并且速度更快2-4倍,还可以注意到,AF也没有接受过蛋白质组装的训练,从而支持了该方法的鲁棒性。
结论表明,只要子组件准确,就可以组装复合物。复合物的对称性会影响结果,并且某些对称性 (二面体和环状) 比其他对称性 (螺旋,不对称) 更丰富且更易于预测。在该计算平台(两个NVIDIA A100,40Gb RAM)上AF (和AFM) 的极限是大致3000个残基,并且所有复合物的73/175 (42%) 都大于该极限。
总而言之,仅使用蛋白质序列信息和化学计量就可以组装具有不同对称性的大型复合物。对零件中的大型复合物进行建模并进行组装,将预测大型复合物的问题转化为对其子组件的预测。这表明了一个令人兴奋的未来,可以对整个细胞中所有蛋白质复合物的模型进行建模。
使用此处提出的方法预测蛋白质复合物的一个局限性是化学计量。通常不知道给定复合物中有多少个蛋白质拷贝,这是组装的要求。一旦通过对复合物的计算或实验研究克服了这一限制,就有可能以新颖的构型组装许多不同的蛋白质复合物。
参考资料 Bryant, P., Pozzati, G., Zhu, W. et al. Predicting the structure of large protein complexes using AlphaFold and Monte Carlo tree search. Nat Commun 13, 6028 (2022). https://doi.org/10.1038/s41467-022-33729-4.

