
编译 | 程昭龙 审稿 | 林荣鑫,王静 本文介绍由美国加利福尼亚州帕萨迪纳加州理工学院生物与生物工程系的Matt Thomson通讯发表在 Nature Computational Science 的研究成果:目前,测序成本是导致单细胞mRNA-seq无法应用于许多生物学和临床分析的主要原因。靶向单细胞mRNA-seq通过分析缩减的基因集来降低测序成本,这些基因集以最少的基因捕获生物信息。为此,作者提出了一种主动学习方法,该方法可以识别数量最少但信息量很大的基因集,从而能够使用少量基因识别单细胞数据中的细胞类型、生理状态和遗传扰动。其中的主动特征选择过程通过使用主动支持向量机 (ActiveSVM) 分类器从单细胞数据中生成最小基因集。经实验证明,ActiveSVM 特征选择识别的基因集在细胞图谱和疾病特征数据集上的细胞类型分类准确率能达到约90%。数量少但信息量大的基因集的发现有助于减少将单细胞 mRNA-seq 应用于临床测试、治疗发现和遗传筛选所需的测量次数。

1 简介 单细胞 mRNA-seq方法的规模已扩大到每次实验可以对数千个细胞进行常规的转录组水平分析。尽管单细胞 mRNA-seq 方法可以为许多不同的生物学和生物医学问题提供见解,但高昂的测序成本阻碍了单细胞 mRNA-seq 在许多探索性分析和成本敏感的临床分析中的广泛应用。靶向 mRNA-seq 的开发有助于解决上述测序瓶颈,通过将测序资源集中在特定生物学问题或分析的高信息量基因上,可将测序成本降低多达 90%。
细胞通过调控转录程序或模块来调控基因表达,这些转录程序或模块包含受共同转录因子组调控的多个基因。由于协同调控,转录模块内的基因表现出相关的基因表达。基因表达的相关性可以使细胞的转录状态能够通过对少量高信息量基因的靶向mRNA分析来重建。然而,这种靶向测序方法需要计算方法来识别针对特定生物学问题的高信息量基因。差异基因表达分析和主成分分析(PCA)等一系列计算方法可用于识别高信息量基因。目前,定义最小基因集的方法在计算上非常昂贵,难以应用于大型单细胞mRNA-seq数据集,并且通常需要用户定义的阈值来进行基因选择。
受主动学习方法的启发,作者开发了一种计算方法,该方法通过主动支持向量机(ActiveSVM)分类任务选择能够可靠识别细胞类型和转录状态的最小基因集。ActiveSVM 算法通过迭代细胞状态分类任务构建最小基因集。在每次迭代中,ActiveSVM 应用当前基因集将细胞分类到类中,这些类由细胞状态的无监督聚类获得或实验标签提供。该程序分析当前基因集错误分类的细胞,然后识别其中最大信息量的基因,并将这些基因添加到正在增长的基因集中以改善分类。ActiveSVM 通过主动查询 SVM 分类器的输出,找出分类不佳的细胞,然后对错误分类的细胞进行详细分析,以选择信息量最大的基因。通过定义明确的分类任务选择最小的基因集,可以确保 ActiveSVM 发现的基因集保留了生物信息。
ActiveSVM 的主要贡献是:该方法可以扩展到超过一百万个细胞的大型单细胞数据集上,这是因为该方法将计算资源集中在分类较差的细胞上。由于该算法仅分析与当前基因集分类较差的细胞的完整转录组,因此该方法可用于发现能够以高精度区分细胞类型的小型基因集,即使在超过一百万个细胞的数据集中也是如此。经实验证明, ActiveSVM 可以在短短数小时内分析包含 130 万个细胞的小鼠大脑数据集。此外,ActiveSVM还可以推广到一系列单细胞数据分析任务中,包括识别疾病标志物、Cas9扰动应答基因和空间转录组学中的区域特异性基因。
2 结果 ActiveSVM特征选择概述 作者开发了一种应用支持向量机分类器来识别小型基因集的计算方法,以区分单细胞数据中的细胞状态(图 1)。该算法通过训练SVM模型,根据标签对细胞类型进行分类,从而迭代的选择基因并使用已识别的基因对细胞进行分类。该算法在给定当前基因集的情况下,识别数据集中分类较差的细胞,并通过分类错误的细胞选择额外的基因,以提高整个数据集的分类精度。
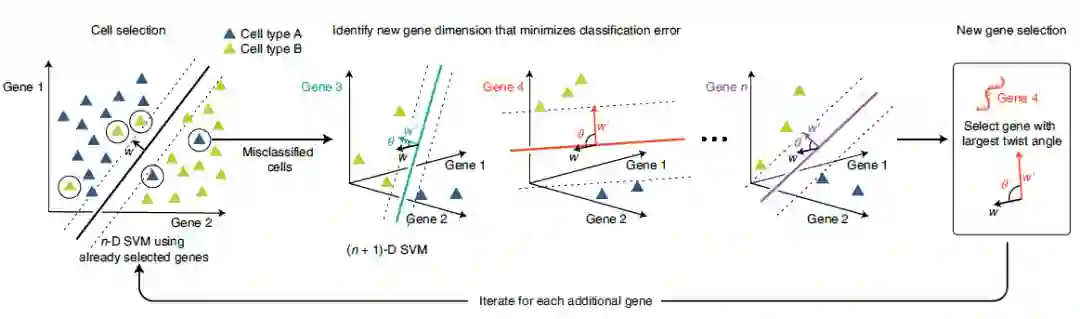
图1 ActiveSVM特征选择概述
ActiveSVM基于一组细胞标签,通过迭代分类和基因选择来构造最小基因集。由于ActiveSVM仅对当前基因集分类较差的细胞执行全转录组分析,因此大大提高了该算法的计算效率。ActiveSVM可以从无监督分析、实验元数据或细胞类型标记基因的生物学知识中获得细胞标签。作者分别提供了最小复杂度和最小细胞版本的 ActiveSVM 算法。最小复杂度算法对一定数量的错误分类细胞进行采样,并直接将其作为细胞集来选择下一个基因。最小细胞算法重用在先前迭代中选择的错误分类细胞,以减少所需细胞的总数。
用ActiveSVM识别单细胞mRNA-seq数据中的最小基因集 作者在四个单细胞 mRNA-seq 数据集(PBMC 数据集、130 万个小鼠大脑细胞数据集、Tabula Muris 小鼠组织数据集和多发性骨髓瘤人类疾病数据集)上测试了ActiveSVM 特征选择方法。在每次分析过程中,显示测试集的分类准确度和选择的基因数量,并将分类性能与几种广泛使用的特征选择方法进行比较,结果表明,ActiveSVM 获得的准确度最高。此外,ActiveSVM 大大减少了时间和内存消耗,特别是对于大型数据集。在与ActiveSVM方法使用相同数量的细胞情况下,所有的比较方法都是逐一选择基因,并根据相应评估函数选择得分最高的新基因。但是,这些方法在每次迭代过程中随机采样细胞,而没有采取主动学习方法。
在人类PBMC数据上的主动特征选择 为了测试 ActiveSVM 的性能,作者将该方法用于提取人类 PBMC 的分类基因子集,分析了包含 6915 个基因的 10194 个细胞的单细胞转录谱数据集,并使用 Louvain 聚类来识别 T 细胞、活化的 T细胞和 NK 细胞、B 细胞和单核细胞。
最小细胞和最小复杂度策略确定的基因组都能以超过85%的准确度对五种主要细胞类型进行分类,且总基因少至15个 (图2a-c)。除了支持数据集的细胞类型分类外,ActiveSVM基因集还提供了一个低维空间来分析数据。主动学习策略的一个关键优点是分析数据集中相对较小的部分,因此该程序可以在只分析298个细胞的情况下生成基因集(图2d)。此外,ActiveSVM可以生成包含已知标记的基因集,标记基因通常对单个细胞类型具有高度特异性,但有些也标记多种细胞类型。
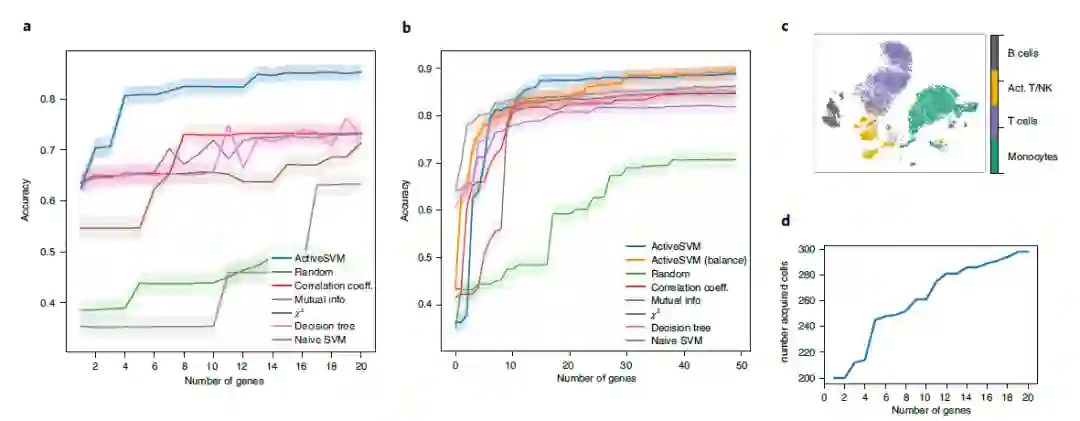
图2 PBMC数据集的基因选择和细胞类型分类
将 ActiveSVM 扩展到百万细胞的小鼠大脑数据集 为了证明ActiveSVM特征选择方法对大型单细胞mRNA-seq数据集的扩展性,作者应用该方法从由10x Genomics收集的大规模细胞演示数据集中提取小型基因集。该数据集包含来自第18天胚胎发育中小鼠大脑的130万个细胞的完整转录组mRNA-seq 数据,它是目前可用的最大的单细胞 mRNA-seq 数据集之一。
实验结果表明,ActiveSVM 分析130万个细胞所需的时间和内存远远小于其他方法。在大规模细胞数据集上,ActiveSVM 发现在分析不到 1000 个细胞时,仅用 50 个基因就可达到约 90% 分类准确度(图 3a-c),ActiveSVM还发现了一系列细胞状态特异性标记基因,扩展了先前的分析(图3d-f)。总的来说,对小鼠大脑细胞数据集的分析表明,ActiveSVM 可扩展到分析超过 100 万个细胞的大型数据集。
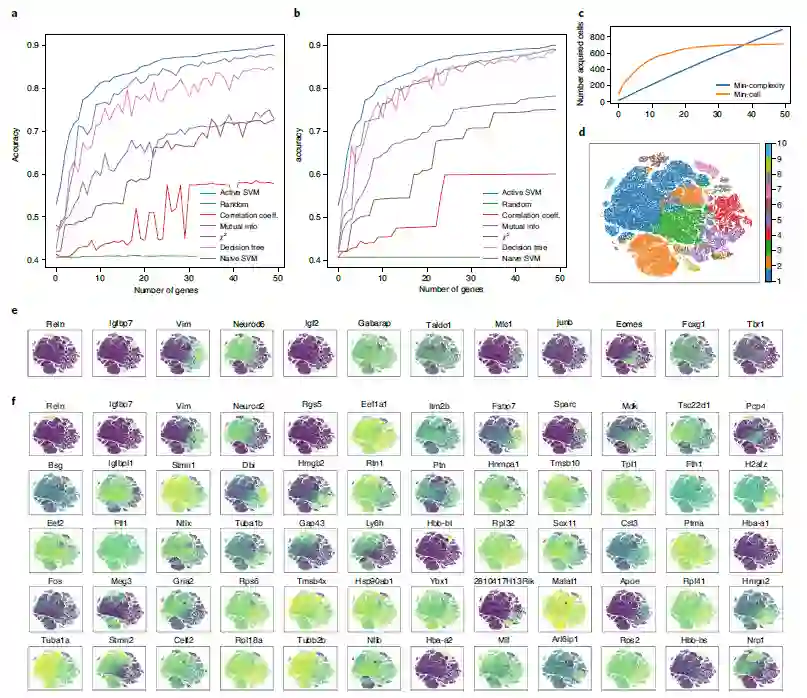
图3 ActiveSVM特征选择扩展到130万个细胞的小鼠大脑数据集
用于小鼠组织研究中细胞类型分类的基因集 除了分析具有大量细胞的数据集外,作者还在具有大量不同细胞类型的数据集上对 ActiveSVM 的特征选择性能进行基准测试。作者将 ActiveSVM 应用于 Tabula Muris 小鼠组织数据集,该数据集包含 58 种带注释的细胞类型和 12 个主要组织的 55656 个单细胞。对于每个细胞,测量 8661 个基因。并且在分析中使用了提供的细胞类型标签,这些标签与组织类型无关。
与其他方法相比,即使有大量的细胞类型, ActiveSVM 也可以构建高精度(>90%)的基因集(图 4a)。为了构建一个大小为500的基因集,ActiveSVM特征选择使用不到800个独特的细胞或者平均每个细胞类型14个细胞。当分析由选定的150个基因(图4c、d)或500个基因组成的低维t-SNE空间内的细胞时,可以从原始数据(图4b)中重建聚类模式。ActiveSVM 能够构建一组识别小鼠不同组织的细胞类型的标记基因,即使在分析大量细胞类型时,也能够识别出细胞类型高度特异性的基因。
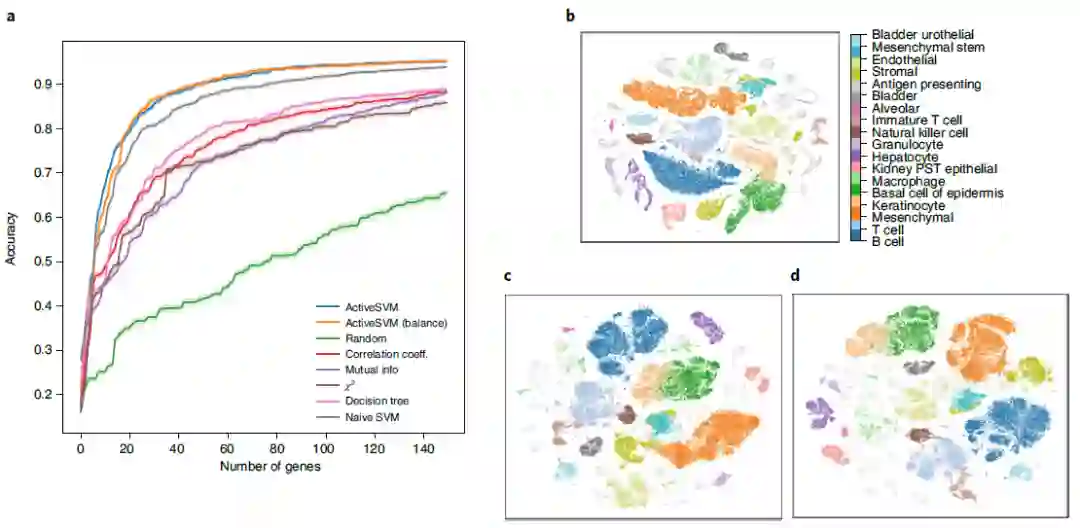
图4 Tabula Muris小鼠组织研究中细胞类型分类的最小基因集
识别多发性骨髓瘤患者的最小基因集 为了分析作为发现疾病特异性标志物工具的ActiveSVM,作者使用了从两名健康供体和四名被诊断为多发性骨髓瘤(一种无法治愈的浆细胞癌)患者的外周血免疫细胞中收集的单细胞数据,该数据集包含35159个细胞和32527个基因。
作者将ActiveSVM的分类精度与其他方法(图5a)进行了比较,发现ActiveSVM在有限的步骤内实现了高精度,并始终优于使用随机和平衡采样的其他方法。在 t-SNE 投影的原始数据集中,确定了健康和多发性骨髓瘤细胞的非重叠细胞类型簇(图 5b)。使用最小复杂度(图5c、d)和最小细胞策略,从40个基因构建的t-SNEs中复制非重叠簇。在数据的t-SNE表示中,使用最小复杂度策略(有或没有细胞平衡),最小的基因集足以将多发性骨髓瘤从健康样本中分离出来。ActiveSVM还识别了外周血免疫细胞内多发性骨髓瘤的已知的和标记的成分(图5e)。结果表明,ActiveSVM可以自动定义与疾病进展和治疗结果有临床关联的基因组。ActiveSVM生成的最小基因集可以为各种临床任务提供有用的靶向测序面板。
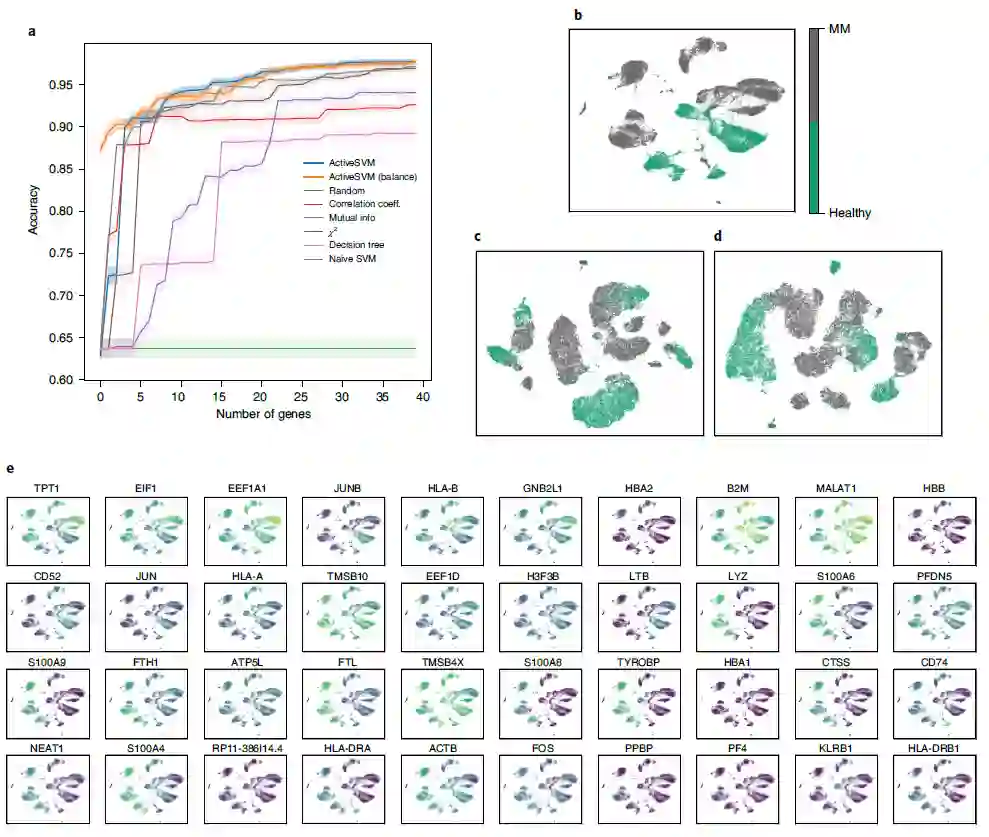
图5 多发性骨髓瘤数据集中健康与疾病分类的基因集选择
识别受Cas9扰动影响的基因 上述分析表明,ActiveSVM在一系列单细胞mRNA-seq数据集中识别了用于细胞状态识别的最小基因集。为了证明基于 ActiveSVM 的基因集选择在单细胞基因组学任务中的泛化能力,作者在另外两个应用中用该方法来识别标记基因:perturb-seq和空间转录组学。
Perturb-seq 是一种使用单细胞 mRNA-seq 读数进行基于 Cas9 的遗传筛选方法,它的优点是可以同时进行多个敲除实验。然而,由于测量和实验噪声,识别遗传扰动对细胞群的影响可能具有挑战性,并且Cas9分子对基因组的切割是不完整的,现在已经开发了各种方法来增强信号。
作者应用ActiveSVM从具有转录因子敲除的小鼠树突状细胞收集的perturb-seq数据中识别了最小的基因集以及转录因子敲除的下游效应。ActiveSVM通过类别平衡策略在Cebp sgRNA细胞标记上识别出最小基因集(50个基因),分类准确率约为80%。ActiveSVM只使用了一小部分数据,而比较方法在整个数据集上执行, ActiveSVM在该数据集上(有噪声)的表现也比其他方法更好(图6a,b)。我们对 perturb-seq 数据的分析表明,ActiveSVM 可以作为一种有用的工具,用于识别由 perturb-seq 实验调节的基因。因此,ActiveSVM 可以提供一种识别最小基因集的方法,该方法可用于增加 perturb-seq 数据收集的规模。

图6 ActiveSVM在perturb-seq 数据上的分类结果对比
用空间转录组学定义大脑区域标记物 最后,为了进一步证明 ActiveSVM 方法的普遍性,作者应用该方法来识别最小基因集,以便按空间转录组数据中的空间位置对细胞进行分类。空间转录组学是一种新兴的方法,用于测量单个细胞内的 mRNA 表达,同时保留组织内的空间信息和细胞相似度。
作者应用 ActiveSVM 来识别与小鼠大脑中特定空间位置相关的基因,并确定了小于30个基因的基因集,这些基因集能够以最小复杂度策略实现位置分类,准确率高于85%。ActiveSVM在每次迭代中仅使用十个细胞,但效果优于在整个数据集上执行的比较方法。空间分析表明,ActiveSVM 能够自动识别富含不同大脑区域的基因。
3 总结 在本文中,作者介绍了一种特征选择方法ActiveSVM,用于在大型单细胞 mRNA-seq 数据集中发现最小基因集。ActiveSVM 通过迭代细胞状态分类策略提取最小基因集,并专门选择位于 SVM 分类器边缘的细胞,然后使用这些分类较差的细胞来搜索信息量最大的基因(特征)。
在生物学上,最近的一项研究强调了转录组中存在的低维结构,当细胞通过包含大量基因的基因表达程序或模块来调节其生理状态时,该结构将出现在基因表达数据中。由于转录模块中的基因表达具有高度相关性,对少量高信息量的特征基因进行测量足以推断细胞的状态。低维结构可以用来降低测量和分析成本,因为必须测量一小部分转录组来推断细胞状态。而作者开发的ActiveSVM作为一种可扩展策略,可用于在细胞状态分类中提取高信息量的基因。
ActiveSVM 方法在当前实践中有一些限制。首先,作者使用单一分类方法(支持向量机)作为计算引擎开发了 ActiveSVM,而主动学习方法可以更广泛地应用于其他分类策略。其次,该方法目前应用监督学习任务(细胞状态分类)来构建最小基因集,在没有明确细胞状态标签的数据集中,可以从无监督的数据聚类中获得标签。主动采样策略可以扩展到更广泛的应用,包括完全无监督的分析方法和微分轨迹分析。第三,在当前的实践中,ActiveSVM 在每一轮中只选择单个基因。而在某些情况下,可能存在信息量很大的基因对或三元组,这些基因对或三元组只能通过明确的组合策略来发现,这些策略可以在每次迭代中搜索提高分类准确性的基因组合。
虽然 ActiveSVM 目前关注的是降低计算成本,但作者希望未来可将主动采样策略直接应用于测量点。在基因组学中,测量资源通常会限制数据采集的规模。单细胞 mRNA-seq 测量目前受到测序和试剂成本的限制,同样,空间基因组学方法也受到成像时间的限制。在未来的工作中,作者的目标是开发能够通过主动采样提高单细胞数据在线采集的策略。通过仅对符合标准的细胞进行测序或成像,可以在测量点实施主动策略。更广泛地说,通过设计实验扰动,实际诱导生物系统产生信息高度丰富的示例,可能会增加测量的信息量。 参考资料 Chen, X., Chen, S. & Thomson, M. Minimal gene set discovery in single-cell mRNA-seq datasets with ActiveSVM. Nat Comput Sci 2, 387–398 (2022). https://doi.org/10.1038/s43588-022-00263-8
数据 http://support.10xgenomics.com/single-cell/datasets https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM2396856 https://github.com/CaiGroup/seqFISH-PLUS
