
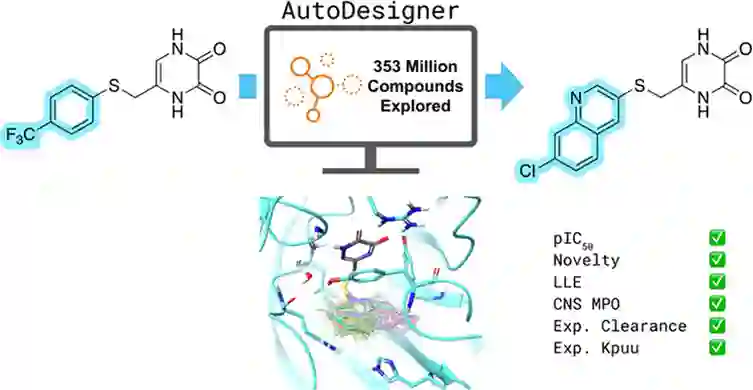
在药物研发过程中,从先导化合物的发现到临床候选药物之间存在着巨大的鸿沟,前者往往只需要具有良好的靶点结合活性及可改造的结构片段,但后者还需要对其药代动力学性质等提出更高的要求。因此,先导化合物优化(Lead optimization)归根结底是一类多参数优化的问题。在现实世界的先导化合物优化研究中,人们关注的更多还是提高小分子在靶点和细胞水平上的亲合力,这一导向往往会使得改造后的化合物亲脂性相对较高,与其他成药性质存在一定的冲突。另一方面,为了实现不同层面上多种分子性质的理想平衡,需要对先导化合物进行反复地改造和优化,从潜在的化学空间中找出满足多方面需求的结构。然而,在项目早期缺乏构效关系与结构数据的情况下,即使是资深的药物化学家也难以从无比广阔的化学空间中设计出值得优先进行测试的分子。 薛定谔公司的Sathesh Bhat等人发展了一种从头设计算法AutoDesigner,通过模仿药物化学家的先导化合物优化思路,广泛地探索符合良好药动性质条件的化学空间,最终设计出新颖、类药且具有理想活性的化合物,并将其成功应用在D-氨基酸氧化酶(DAO)抑制剂的设计项目当中,大大加速了先导化合物优化的过程。这项工作最近发表在美国化学会出版的计算化学和化学信息学核心期刊Journal of Chemical Information and Modeling上(J. Chem. Inf. Model. 2022, 62, 1905-1915)【1】。
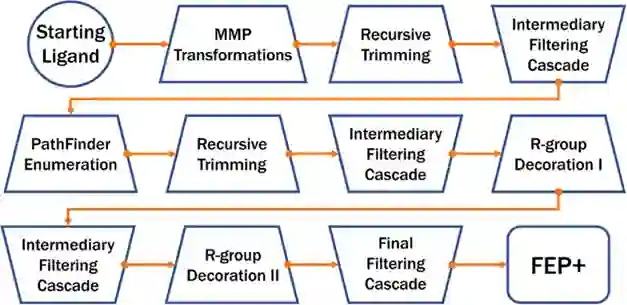
图1:AutoDesigner算法的工作流示意图 从整体上看,AutoDesigner算法的每一轮运行都会经历三种生成分子以及紧随其后的过滤筛选阶段(如图1)。其中,分子生成的机制可以具体分为匹配分子对转换(MMP Transformations)、基于反应的枚举(PathFinder enumeration)、递归结构修剪(Recursive Trimming)以及R基团修饰(R-group Decoration),过滤筛选阶段也可以分作中间环节的筛选以及最终给出结果的筛选两种情况。 匹配分子对转换这一机制依赖于从PubChem和ChEMBL数据库中获取得到的海量匹配分子对,通过将两个数据库中的分子按照预定义的规则进行碎片化,再组合到片段间的转化上,分别能得到约2.91亿、0.26亿种结构转换。值得指出的是,在对输入的配体分子进行转换之前,会优先进行递归结构修剪,从而在一开始将输入的单个配体转变成一组结构相似的化合物(如图2),有效扩大了分子对转换时的化学空间覆盖。此外,不论哪种分子生成机制,都不改变预定义的核心结构,如图2中蓝色发光标记的片段。
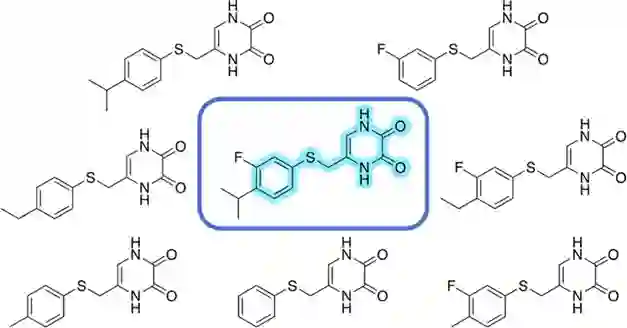
图2:对方框中分子进行递归结构修剪的结果展示 如果说匹配分子对转换模仿的是药物化学家基于原始结构进行的衍生与改造,那么基于反应的枚举与R基团修饰则更加体现了组合化学的思想,参考药物化学家在设计目标分子的合成路线时,对各个反应步骤所使用的不同反应物进行组合。AutoDesigner算法采用薛定谔公司此前报道的PathFinder技术,对所给分子进行逆合成分析,并对反应物中所有可以购买的原料进行组合化学枚举。一般而言,在目标化合物的最后一步合成中,人们会大量改变所使用的试剂或原料,从而向骨架中引入丰富多样的修饰基团。类似地,AutoDesigner利用精选的R基团数据库,向所给分子中每一个可改造的位点进行修饰,对于那些经过一轮修饰后还有修饰空间的分子,还会进行第二轮的修饰。通过这两种分子生成机制,该算法在有效实现分子结构多样性的同时,兼顾了可合成性。 四种分子生成机制保证AutoDesigner算法能充分地搜索丰富多样性的化学结构,而有效的过滤筛选机制则是实现多参数优化的重要工具。中间环节的过滤筛选需要在保留有潜力的化合物前提下筛除不需要的化合物,因此设置的筛选条件较少,并能让那些在后续改造过程中有机会保留下来的分子不被排除,例如不对氢键供体的数量设置筛选条件。相对的,最终环节的过滤筛选则尽可能使得条件严格,仅允许满足项目目标化合物性质的分子得以通过。值得注意的是,筛选条件的制订不拘泥于经典的五倍率,而是由药物化学家根据项目本身需要进行灵活的调整。此外,两种不同强度的筛选环节也节约了算法整体所消耗的计算资源。 为了实现对生成分子各项性质的筛选,作者为AutoDesigner算法准备了用于计算亲合力与药代性质的计算工具。前者通过薛定谔公司的FEP+模块辅以主动学习技术实现加速优化,后者则通过机器学习模型AutoQSAR预测药代性质。最后,AutoDesigner算法还被部署到云计算平台上,从而提高对计算资源的调配水平,提高效率。 利用这一先导化合物优化工作流,作者依次运行了三个阶段来对DAO抑制剂先导化合物进行优化(图3),其输入配体分别为化合物1,5,7。化合物2~4、化合物6及化合物8依次属于三个阶段优化输出的分子,如图3所示。结果表明,经过AutoDesigner的优化后,这些分子都能保持较为理想的抑制活性,而其他方面的干湿实验也表明在众多理化性质和药代性质上有所改进,这意味着该算法工作流具有出色的实战价值,能够较好达成其预期目标。
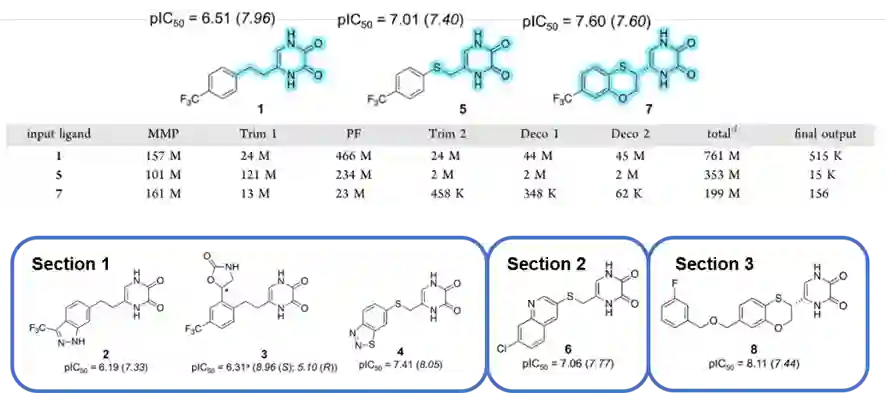
图3:三个阶段中AutoDesigner各环节生成的分子数目与起始或代表性DAO抑制剂的结构、活性数据(括号中为预测值) 为了进一步探索DAO结合部位的性质,作者应用AutoDesigner完成了一项先导化合物优化的挑战任务。在此前的文献报道中,由于对DAO抑制剂结合口袋的了解不深,因此认为口袋的形状大小较为狭窄有限,向先导化合物中引入较大的修饰基团时倾向于降低抑制活性。因此作者以化合物7作为这一阶段算法运行的输入,探索DAO抑制剂结合口袋的空间,尝试向分子中的疏水芳基尾部引入基团来进一步提高抑制活性。
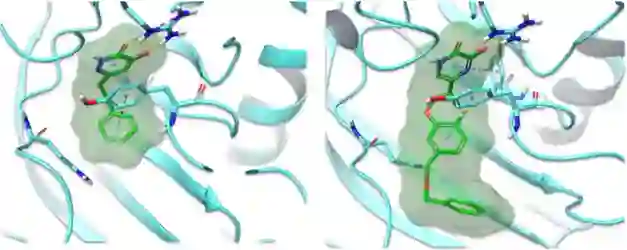
图4:化合物7、8在靶点口袋中的占据情况比较 从图3中可以发现,AutoDesigner共生成近2亿个分子,但最终仅有156个符合理化、生物活性及药代等各方面的性质要求,可见这一任务的困难程度。其中,化合物8是这一批优化结果中的代表分子,如图4所示,其结构中延伸出的氟代苯基进入到了DAO结合口袋深处。这样的结构改造使其成为该项目中得到的抑制活性最强的分子。从这一实例可以看出,AutoDesigner能够有效识别出配体与靶点之间的已知构效关系,甚至可以辅助药物化学家进一步探索未知的构效关系,从而助力先导化合物的优化环节。小 结
这项工作报道的AutoDesigner通过广度的搜索算法辅以云计算,成功对庞大的化学空间进行了连续多轮的探索和过滤,并且基于项目本身需求配合上药物化学家经验指导的过滤条件设置,使得分子改造结果可以满足多参数优化的需要。这个算法在DAO抑制剂设计的应用实践中,彰显了AutoDesigner的实用价值与可靠性,其能够在缺乏大量过往实验数据积累的情况下,共生成并筛选了超过10亿种化合物,分析出其中的重要相互作用,甚至能够发现新的构效关系。这意味着AutoDesigner不仅可以为药物化学家确定化合物合成、测试的优先顺序提供参考,还能够协助人们进一步探索未被发现的结构信息。这项工作表明利用算法模拟传统药物化学的研究思路,或能确保计算方法的稳健性与可靠性。与此同时,随着算力和计算精度的逐步提高,应用于药物设计与研发的计算工具正逐渐从实验室模拟向真实世界应用的方向不断发展。
参考文献【1】Bos, P. H.; Houang, E. M.; Ranalli, F.; Leffler, A. E.; Boyles, N. A.; Eyrich, V. A.; Luria, Y.; Katz, D.; Tang, H.; Abel, R.; Bhat, S., AutoDesigner, a De Novo Design Algorithm for Rapidly Exploring Large Chemical Space for Lead Optimization: Application to the Design and Synthesis of d-Amino Acid Oxidase Inhibitors. J. Chem. Inf. Model. 2022, 62, 1905−1915. (DOI: 10.1021/acs.jcim.2c00072)
