新药物设计是产生具有理想药理和理化性质的新型先导化合物的过程。深度学习(deep learning, DL)在药物从头设计中的应用已成为一个热门话题,许多基于深度学习的方法已被开发用于分子生成任务。一般来说,这些方法按照四个框架开发:循环神经网络;编码器-解码器;强化学习;以及生成式对抗网络。在这篇综述中,我们首先介绍了基于深度学习的新药从头设计中使用的分子表征和评估指标。然后,我们总结了每种架构的特点。最后,对基于深度学习的分子生成技术的潜在挑战和未来发展方向进行了展望。
https://www.sciencedirect.com/science/article/pii/S0959440X21001433?via%3Dihub
据估计,化学空间中类药物化合物的数量可能高达1023-1060[1],因此完全探索这一巨大的化学空间在计算上是非常困难的。在这种情况下,如何有效地从如此大的化学空间中挖掘新的先导化合物成为药物发现的一个真正的挑战。随着计算能力和实验技术的显著提高,高通量筛选[2]和虚拟筛选(VS)[3]已成功应用于多种过滤器对大型化学库进行评价。此外,随着机器学习(ML)技术的快速发展,建立了基于ML的定量构效关系(QSAR)方法,作为VS不可缺少的过滤器,有助于高效、可靠地评估各种理化和药理特性[4]。然而,传统的基于QSAR或基于对接的VS通常倾向于从现有的化学库中寻找具有理想性质的分子。相比之下,新药发现(de novo drug design,分子生成)旨在通过从头生成具有理想性质的新分子来补充现有的化学库,从而探索化学空间。
尽管许多基于计算生长算法和进化算法的传统从头药物设计方法已被开发出来,以从构建模块[5]生成新的分子结构,但在许多方法中,优化各种目标和生成新的化合物[5]之间存在折衷。深度学习(DL)的发展为创新药物的设计和发现提供了新的机遇。近年来,开发了各种基于DL的从头药物设计算法,DL在药物发现中的成功应用被麻省理工学院(MIT) Technology Review评选为2020[6]十大突破性技术之一。
在VS中实现的基于深度学习的方法通常用于预测输入分子的物理化学或生物性质,这实际上是QSAR判别建模的应用。与DL在VS中的区别作用相比,基于DL的生成模型本质上可以被认为是在巨大的化学空间中检测具有理想性质的化合物的探索者。基于dl的生成模型可以实现对化学空间中现有分子的性质和/或结构特征进行总结和提取,然后将其转化为新的支架的过程,也称为反向QSAR过程[7]。生成模型的目的是有点类似于它与优化策略适用于接近理想的分子性质(如右所示图1)上角,而与通常方法实现各种过滤器来缩小筛选化合物的化学空间,直到达到一个容易处理的范围(如右所示低的图1)。不过,相信,通过探索的连续空间的属性,生成模型可以生成具有新颖支架和理想性质的分子。
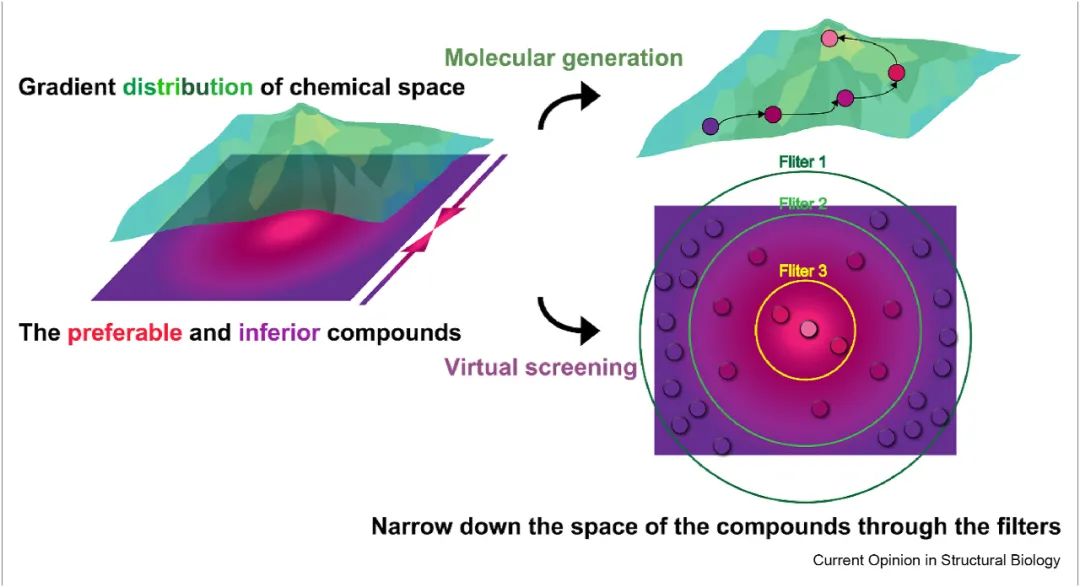
图1 通过虚拟筛选和分子生成来探索化学空间的过程。虚拟筛选通过给定的化学库的性质来筛选化合物,而新药设计通过聚焦于定向性质来生成新的化合物。
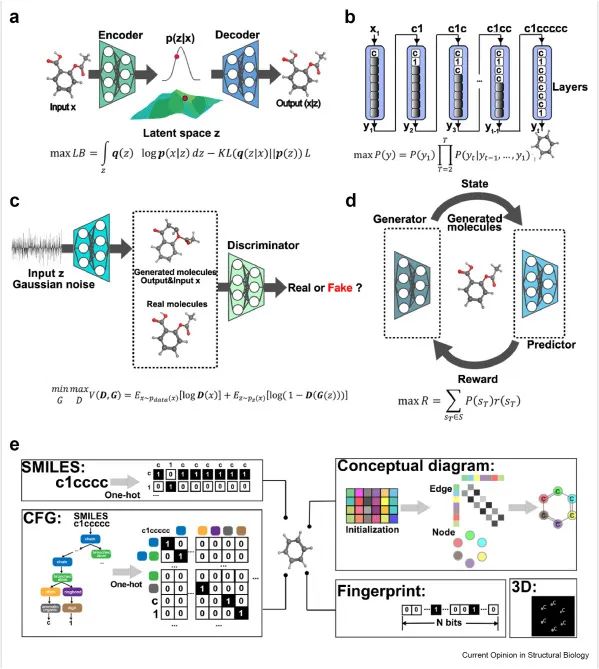
ML算法用于构建生成模型可以分为四类:递归神经网络(RNN) encoder-decoder (Enc-Dec),强化学习(RL)和生成对抗网络(GAN)(8、9)(图2)。尽管传统的生成模型已经开发的各种变异,其中大部分集中在两个基本问题:分子表征;优化策略[8]。不同表示的应用旨在使DL算法更容易理解分子的不同特征。报道的算法除了常用的简化分子输入线输入系统(SMILES)[10,11]外,还使用了分子图[12,13]、指纹[14]和三维(3D)几何[15]。生成过程中对分子性质的优化是为了使生成的分子在化学空间中产生高梯度和聚集区域,具有较高的有效性、新颖性和可合成性。例如,GENTRL[10]应用张量分解和自组织映射技术优化分子在化学空间的性质,快速设计针对Discoidin Domain Receptor Tyrosine Kinase 1 (DDR1)激酶的新型活性化合物。
通过这些改进的算法,大量基于深度学习的模型已成功用于目标导向的分子设计,并通过有机合成和活性评价成功识别了新的活性分子[10,16,17],凸显了基于深度学习的生成模型在从头药物设计中的潜力。本文重点介绍了用于新药设计的各种最新DL架构的特点。