ScienceDirect|AI 在3D化合物设计中的应用综述

现在许多AI算法要么根本不考虑靶标的3D结构,要么难以从靶标中捕获有意义的空间信息。
本文重点介绍一系列考虑结构信息的深度学习方法,这些方法利用深度学习进行化合物设计和虚拟筛选。
本文讨论了如何通过促进符合特定设计假设的化合物的设计,以及通过揭示可用于帮助分子设计的关键蛋白质-配体相关作用,来更好地将这些方法整合到现有的药物发现的管线中。
一、用于化合物设计的深度生成模型
分子设计在很大程度上依赖于药物化学家的专业知识,他们利用对化学空间和靶标结构的理解来设计形成有希望的相互作用的配体。
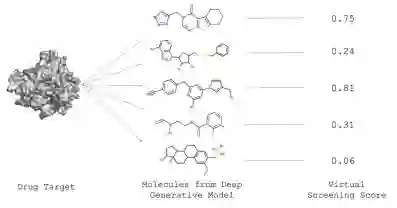
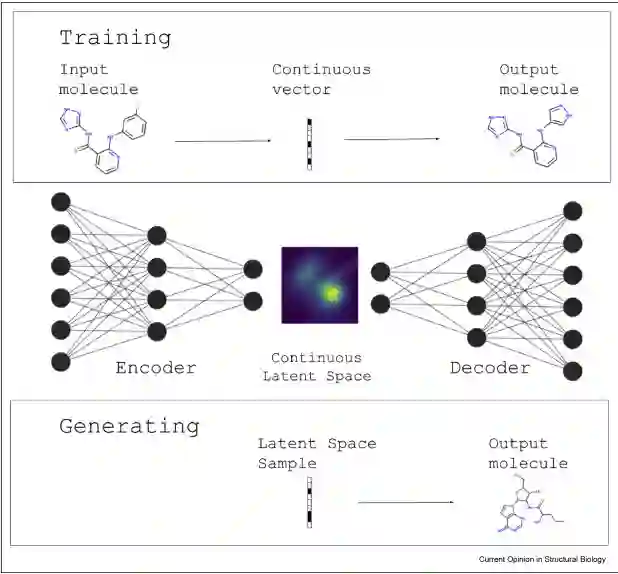
然而,即使可以使用现有的计算技术,例如对接算法和分子动力学模拟,这个过程也是昂贵且耗时的。深度生成模型越来越多地被提议作为分子设计的替代方法。这些方法旨在能够快速生成多个高亲和力结合剂。下图展示了用于药物发现的深度生成模型的示意图
用于分子生成的早期ML模型将分子表示为SMILES字符串,这是一系列完整描述2D化学结构的ASCII字符,但是早期的例子在模型中缺乏语法限制,这样导致生成的很大比例的分子是无效的。
最近的模型建议使用图来表示分子,要么atom by-atom地构建分子,要么从分子构建块的词汇表中构建分子,[1]证明他们的基于图模型生成的分子具有优于基于SNILES的方法的特性。
深度生成模型的一个吸引人的特性是,针对特定的属性优化分子,它们允许通过搜索算法(如贝叶斯优化)或使用强化学习训练模型。[2]试图生成对特定蛋白靶标具有活性的分子,但他们的方法严重依赖于已知对蛋白质具有活性的分子的存在,从而限制了它们在为新靶标设计分子时的适用性。
与需要已知活性库的模型相比,少数方法没有将已知活性作为针对特定靶标设计分子的基础,而是将蛋白质特定的3D信息明确纳入其生成过程,使其适用于范围更广的目标。
第一个使用结构感知的生成模型LiGANN[3]生成了一组与结合口袋互补的配体形状,使用形状描述网络生成具有能够与蛋白质结合的药效团的配体。与从ZINC15中获得的随机样本相比,LiGANN生成的配体在包含31个靶标的测试集上具有优异的对接分数。
[4]还通过将原子密度网格编码进配体和蛋白质的单独隐变量表示中,并训练模型以根据蛋白质结构生成3D配体来整合3D信息。作者表示,与不考虑蛋白质结构的类似模型相比,在生成分子时加入蛋白质结构信息通常会改善所得分子的3D特性。
与上述方法相比,这些方法明确提供了蛋白质结构的表示作为模型的输入,[5]通过将对接分数纳入其奖励函数来激励生成分子,从而在分子生成中利用强化学习以高亲和力结合蛋白质的分子。[6]提出了一种基于Autodock Vina生成具有适应度分数的分子的遗传算法,该算法可以预测蛋白质和配体之间的结合亲和力。
以上方法类似于高通量筛选,一种不同的策略,是在某蛋白质背景下,明确地结合/构建已知分子的粘合剂。Delinker[7]是一个片段连接的生成模型,它以两个片段和描述片段相对位置的3D信息作为输入并返回一个连接的分子。[8]证明了用户指定施加的3D药效团约束对原始Delinker模型产生了实质性改进,并允许对生成的分子的药效团谱进行更大程度的控制,从而更容易集成到符合药物化学家通常制定的设计假设,并试图枚举符合药物研发过程中的那个假设的分子。
深度生成模型的迅速普及意味着用户很难评估它们应该为特定任务使用哪种方法。不同的模型通常使用不同的分子集进行训练和测试,并使用不同的指标进行评估。因此,需要一组基准,使从业者能够轻松评估哪种生成模型最适合他们的设计任务。MOSES和GuacaMol最近发布的平台,可促进在一系列分布学习和靶标导向基准中比较不同生成模型。然而,现有的靶标导向基准侧重于生成与现有配体高度相似的分子,而不是可能与特定蛋白质结合的分子。
二、基于结构的虚拟筛选
特定的分子是否会与感兴趣的蛋白质结合是药物发现中的核心问题。在考虑大量分子时,就像使用生成模型时经常出现的情况一样。重要的是能够廉价而准确地评估哪些分子将与蛋白质结合,以避免合成大量无活性分子。
分子对接工具利用经验或基于知识的评分函数来预测配体的正确结合模式并估计结合亲和力蛋白质-配体复合物。对接协议广泛用于虚拟筛选活动,以区分结合剂和非结合剂,并快速优先考虑来自更大库的少量有希望的分子。
早期机器学习算法将蛋白质-配体相互作用指纹作为输入,并将配体分类为结合剂或非结合剂;虽然这些模型经常被报道为表现出优于经典评分函数的预测准确率,但随后的研究提出了它们对姿势变化缺乏敏感性以及它们泛化到新靶标的能力的担忧。
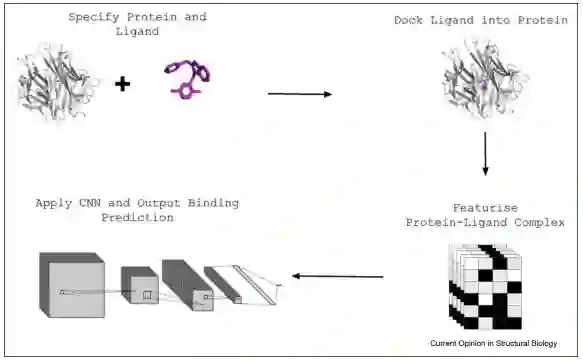
受卷积网络CNN捕获图像识别任务中重要空间信息能力的启发,几位作者试图通过使用对接的蛋白质配体复合物作为CNN的输入来预测蛋白质-配体结合。
虽然蛋白质结构在基于深度学习的化合物设计和蛋白质配体评分方法中发挥着越来越重要的作用,但通常这些模型具有单一的静态结构,限制了模型解释结合位点灵活性的能力。为了解释蛋白质的灵活性,将机器学习算法应用于进行快速分子模拟已经引起了相当大的兴趣。将此类模拟纳入化合物设计和蛋白质配体评分管道将是未来研究的重要途径。
建立对基于结构的虚拟筛选方法的更大信任的一个关键步骤是,开发允许用户了解配体的哪些特征意味着模型预测它会结合的方法(下图显示了一个示例蛋白质-配体复合物,该复合物已被分配了这样的属性)。
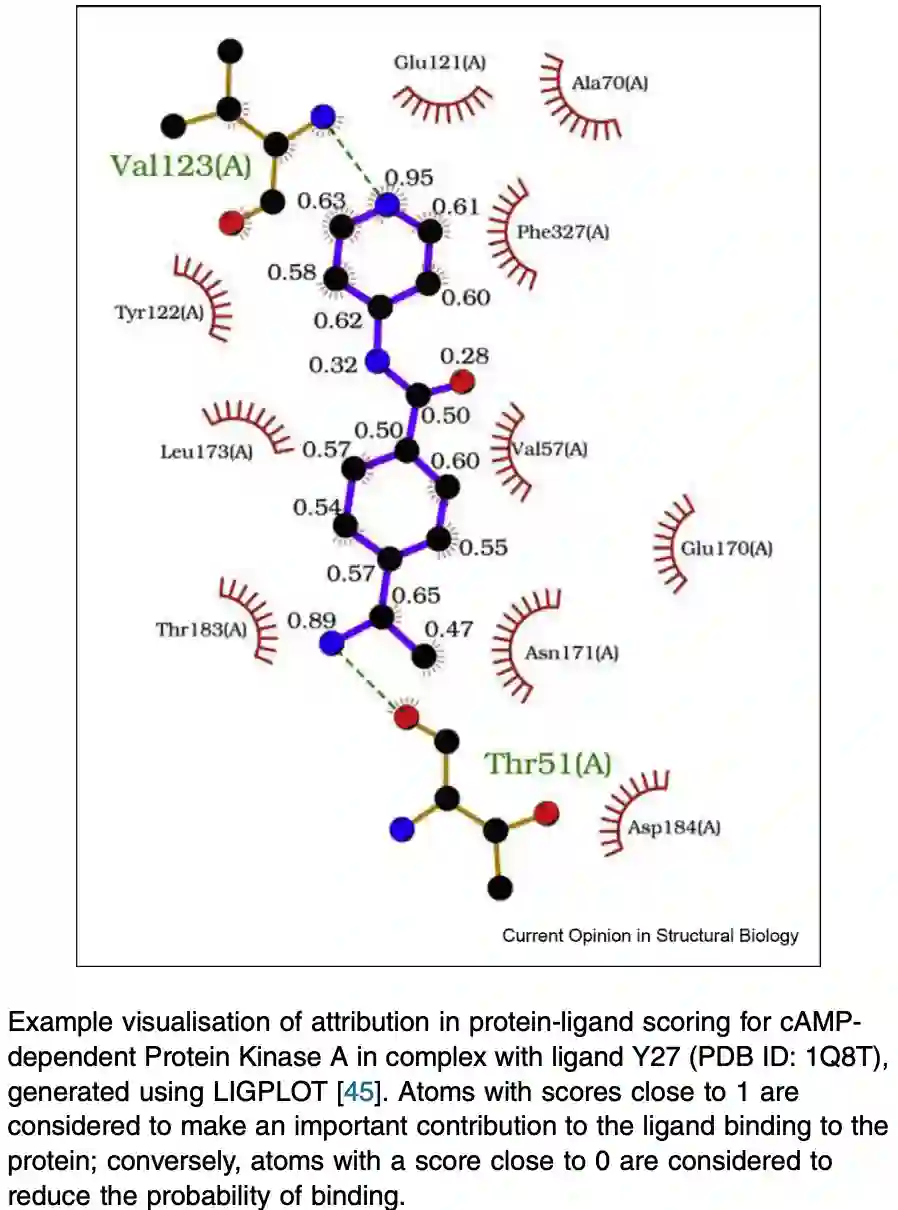
霍楚利等人。[ 9 ] 提出了几种可视化特定原子对 CNN 模型分类的影响的方法,例如通过使用整个蛋白质-配体复合物预测结合概率和使用去除一个原子的整个复合物;然后将两个分数的差异视为该原子对模型绑定决策的“贡献”。布朗等人。
[ 10]提出了一种方法,该方法将同属配体系列作为输入,并预测哪些子结构相对于该系列中的其他配体提高或降低结合亲和力,从而更容易进行迭代细化。
[ 11] 最近提出了一组用于评估归因方法的指标,旨在促进开发更具可解释性的深度学习模型。此类方法的开发是将蛋白质-配体评分模型进一步整合到药物发现活动中的重要一步,因为现有模型目前仅预测特定配体是否会与蛋白质结合,但没有提供关于哪些基序起关键作用的见解在允许绑定发生中的作用;准确的归因方法将更容易让从业者保留重要的基序并丢弃那些无助于配体结合的基序,并可用于告知生成模型制造的分子种类。
三、结论
尽管那些开发生成模型的人长期希望通过输入蛋白质结构并在短时间内接收高效配体来完全自动化药物发现过程,但很明显,在可预见的未来,人类专家仍将是参与分子设计的不可或缺的一部分。因此,生成模型需要易于访问和使用,并且能够定制他们生成的分子以符合一个或多个指定的设计假设。
同样,除了能够区分结合剂和非结合剂外,虚拟筛选模型还需要突出重要的相互作用,这可以让化学家和生成模型开发出更有前途的分子。
朝着这个方向迈出的关键一步是创建大型、无偏见的虚拟筛选数据集,以允许模型学习重要的生物物理相互作用。
参考文献
[1] Jin W, Barzilay R, Jaakkola T: Junction tree variational autoencoder for molecular graph generation
[2] Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol 2019
[3] From target to drug: generative modeling for the multimodal structure-based ligand design. Mol Pharm 2019
[4] Generating 3d molecular structures conditional on a receptor binding site with deep generative models. arXiv preprint arXiv:2010.
[5]Autonomous molecule generation using reinforcement learning and docking to develop potential novel inhibitors. Sci Rep 2020
[6] Molaical: a soft tool for 3d drug design of protein targets by artificial intelligence and classical algorithm. Briefings Bioinf 2021
[7] Deep generative models for 3d linker design. J Chem Inf Model 2020
[8] Deep generative design with 3D pharmacophoric constraints. Chem Sci 2021
[9] Visualizing convolutional neural network protein-ligand scoring. J Mol Graph Model 201808.
[10] General Pur- pose Structure-Based drug discovery neural network score functions with human-interpretable pharmacophore maps. J Chem Inf Model 2021
[11] Evaluating attribution for graph neural networks. In Larochelle H, Ranzato M, Hadsell R, Balcan MF, Lin H. Advances in neural information processing systems, vol. 33. Curran Associates