

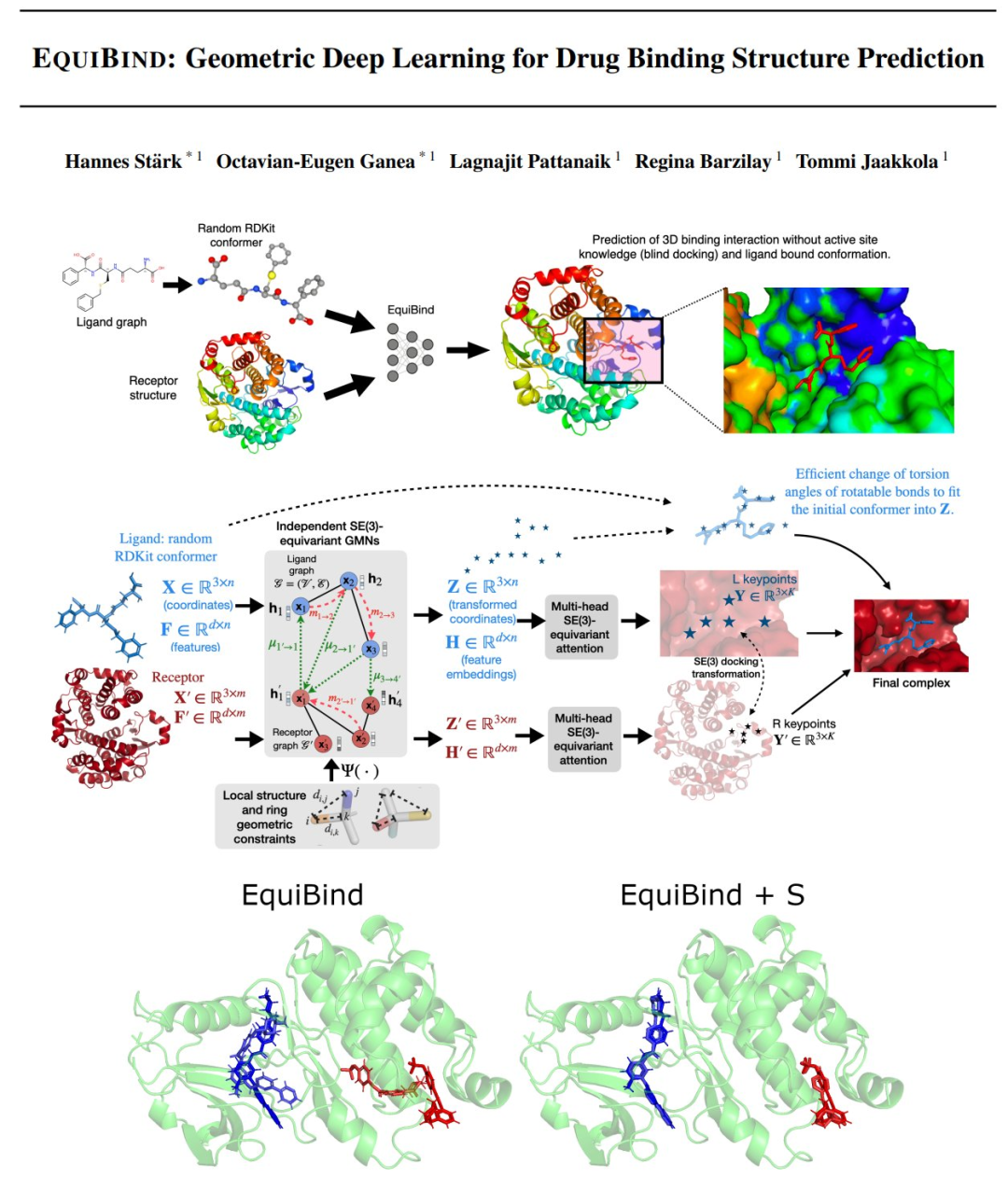
预测类药物分子如何结合到特定的蛋白质目标是药物发现的核心问题。一种极其快速的计算绑定方法将使快速虚拟筛选或药物工程等关键应用成为可能。现有方法的计算成本很高,因为它们依赖于大量的候选样本,并结合了评分、排名和微调步骤。我们用一种SE(3)-等变几何深度学习模型EQUIBIND挑战这一范式,该模型可以直接预测
i)受体结合位置(盲对接)和 ii)配体结合姿势和方向。与传统和最近的基线相比,EquiBind实现了显著的加速和更好的质量。此外,我们还展示了在以增加运行时间为代价将其与现有的微调技术结合使用时的额外改进。最后,我们提出了一种新颖的、快速的微调模型,该模型根据给定输入原子点云的von Mises角距离的封闭全局极小值来调整配体旋转键的扭转角,避免了先前昂贵的能量最小化差分进化策略。
https://www.zhuanzhi.ai/paper/7e1cc60c20e48a58c627b1779b77c957
成为VIP会员查看完整内容
相关内容

Arxiv
0+阅读 · 2022年7月9日
Arxiv
0+阅读 · 2022年7月8日
Arxiv
11+阅读 · 2021年9月24日
Arxiv
21+阅读 · 2021年6月16日

相关VIP内容
相关资讯
相关论文
Arxiv
0+阅读 · 2022年7月9日
Arxiv
0+阅读 · 2022年7月8日
Arxiv
11+阅读 · 2021年9月24日
Arxiv
21+阅读 · 2021年6月16日