

今天给大家介绍韩国江原国立大学Umit V.等人在2022年在Nature Communications发表的一篇名为“Retrosynthetic reaction pathway prediction through neural machine translation of atomic environments”的文章[1]。尽管有机化学的知识已经积累了几十年,但为药物分子设计有效的合成路线仍然是有机合成中的一项关键任务。在这项工作中,作者提出一种新的单步逆合成预测方法:RetroTRAE,即通过学习参与化学反应的原子的环境变化来预测候选反应物。结果显示,在UPSTO测试数据集上的Top-1准确率为58.3%,在相似化合物较多的情况下,准确率为61.6%,优于其他基于神经机器翻译的逆合成方法。同时该方法可有效解决基于SMILES方法的不可解释性以及生成无效字符串等问题研究背景
逆合成设计是有机化学的关键问题之一。现有的逆合成方法可分为基于模板的方法和无模板的方法。其中基于模板的逆合成方法不仅需要克服枚举反应模板而导致的高计算成本,且只能预测模板库中的反应。而无模板方法可有效避免上述的问题,在预测逆合成时表现出更强的泛化能力。无模板方法可进一步细分为基于图神经网络的逆合成预测算法和基于序列的逆合成预测算法。其中,基于序列的逆合成预测算法将反应路线的预测问题看作一项语言翻译任务来处理,将产物的SMILES转化为反应物的SMILES。但目前生成的无效SMILES字符串数量较多,可分为两种类型:(1)语法无效的SMILES字符串;(2)语法有效但语义无效的SMILES。 这项研究通过将反应物的原子环境(Atom Environments, AE)与目标分子相关联,使用原子环境替代传统的SMILES进行单步逆合成预测。该方法使我们通过关注与反应中心相关片段来捕捉化学变化。实验结果表明该算法的性能大大优于现有方法。************模型与方法****************1.模型框架
作者使用原子环境(AE)替代SMILES进行逆合成预测,AE是指以特定原子为中心,不同“半径”的圆形拓扑邻域片段,也包含所涵盖原子之间的所有化学键。其中,“特定原子”称为中心原子,“半径”指的是中心原子和所有共价键原子之间允许的最大拓扑距离。因此半径为 r 的AE包含分子中与中心原子的拓扑距离为 r 或更小的所有原子,以及它们之间的所有键。根据定义,r = 0 的AE只包括中心原子类型的原子,表示为AE0。r = 1的AE包含中心原子、与中心原子相邻的所有原子,以及中心原子与这些原子之间的所有键,表示为AE2。如图1(b)所示,化合物苯的文本描述是以常见的SMILES、SMARTS模式,和新开发的SELFIES模式,以及代表ECFP指纹的AE。 图1(a)提供了该模型的整体流程。首先将产物分解为一组AEs。由SMART模式描述的每个AE都与一个特殊的整数值有关。将AEs列表作为模型RetroTRAE的输入序列,用该模型来预测反应物的AE序列。
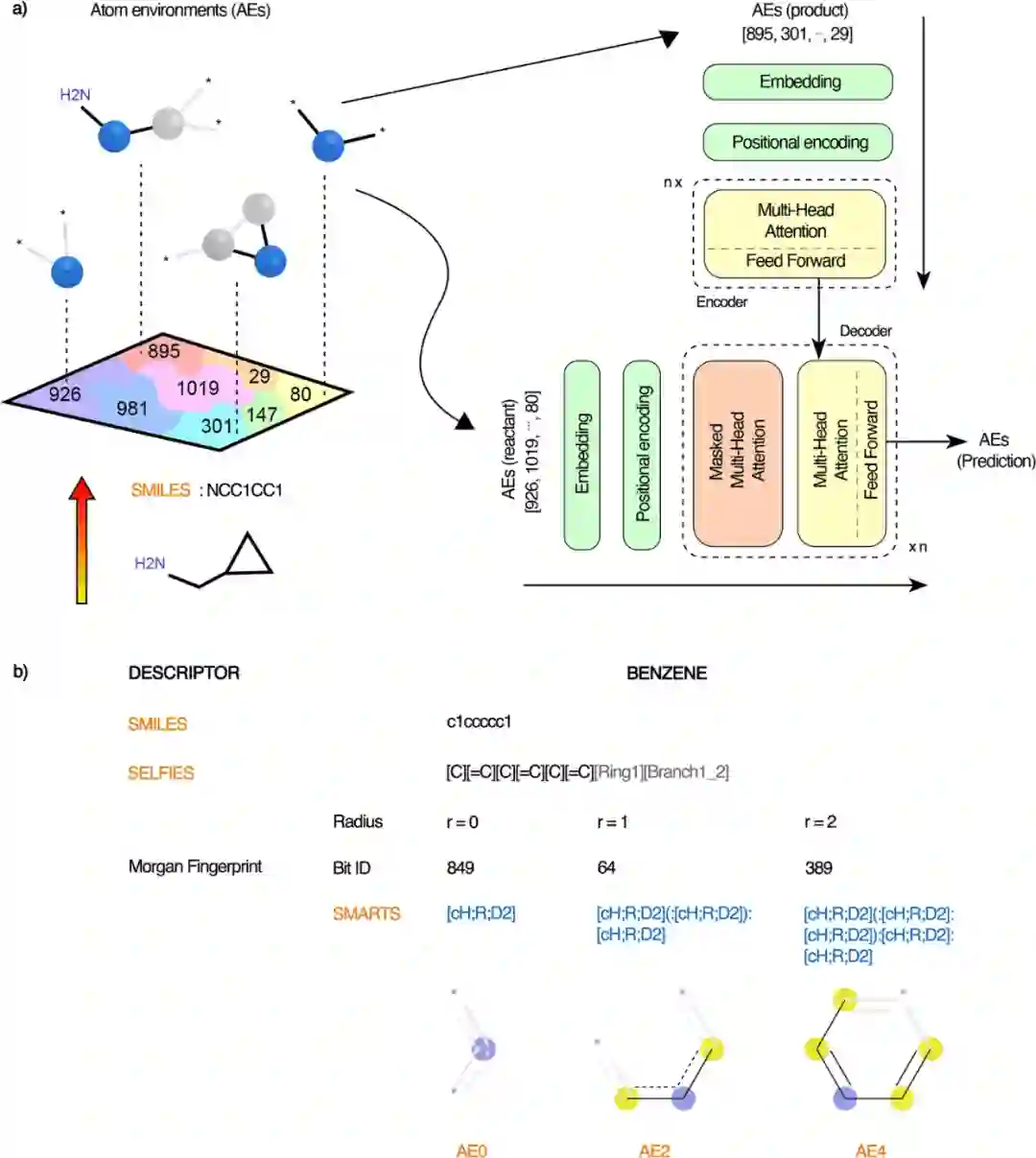
图1 (a):RetroTRAE模型流程图;(b)苯的字符串表示以SMILES、SELFIES以及SMARTS模式。在AEs渲染中,中心原子以蓝色突出,而芳香族和脂肪族环状原子分别以黄色和灰色突出。通配符[*]被用来代表任何原子******[1]**********************2.分子片段比较
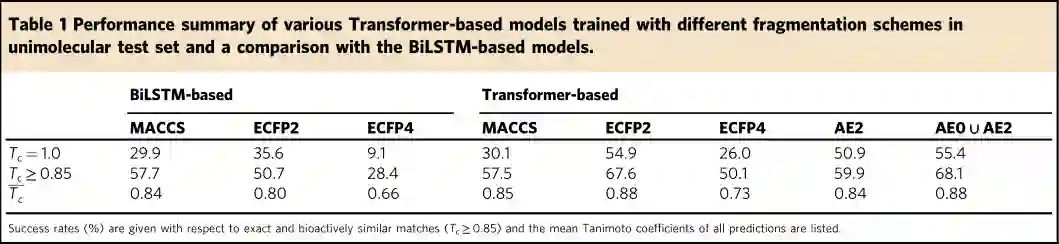
找到一组最准确地代表分子结构的最佳片段是提高逆合成预测性能的关键因素。因此作者使用多个不同的分子片段描述在单反应物测试集中进行逆合成预测评估。如表1所示,作者一共选择了三种分子片段的描述方法进行比较,并分别使用不同的模型框架,性能评价选择衡量相似度的谷本系数(Tanimoto Coefficient, Tc)。 首先,基于Transformer的逆合成模型,在准确性方面与以前基于BiLSTM的方法相比有很大的改进。其次,在MACCS、ECFP2、ECFP4、AE2、AE0∪AE2几种不同的分子片段描述方法中,基于AE0∪AE2的分子Transformer模型性能最佳,达到了55.4%的精确匹配精度。增加生物活性相似的预测(Tc ≥ 0.85)后,准确率也相应提高,模型的总体准确率达到68.1%。因此,作者把具有AE0和AE2联合的Transformer模型命名为RetroTRAE。 表1 在单反应物测试集中使用不同分子片段方案训练的Transformer模型的性能总结以及与基于BiLSTM的模型的比较******[1]**********
**************结果与讨论**************1.RetroTRAE模型性能
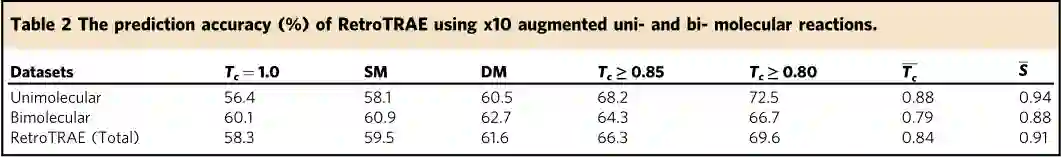
与基于SMILES的方法不同,使用AEs的一个优点是解码不会生成无效或完全不同的分子。模型可解码生成与真实分子高度相似的AEs预测集,为逆合成预测提供有用的信息。 除了采用精准匹配(Tc = 1.0)方式来评估准确率以外,作者在评估模型性能时又增加了四个不同评价节点,四个节点可以分为两类:(a)硬阈值;(b)软阈值。作者将硬阈值定义为单片段(SM)或双片段(DM)差异。反之,将基于Tanimoto系数的任意阈值称为软阈值,如Tc ≥ 0.85,用来筛选具有相似生物活性的分子。作者更强调硬阈值的使用,与软阈值相比,硬阈值(SM/DM)预测分子与真实分子相比,只有某些子结构、官能团等差异,这些小的差异很容易通过与真实分子的视觉比较,找到与真实反应物不同的片段类型和数量,然后进行改正。 作者使用经过过滤的美国专利反映数据集USPTO-full的子集进行模型的性能评估和比较。忽略数据集中的多组分反应,因为此类反应在整个数据集中所占比例不足1.65%。然后根据反应物的数量,最终确定两个不同的数据集,分别包括单反应物(R—>P)和双反应物(R1+R2—>P)类型的反应,大小分别为100 K 和314 K。 此外,作者就使用数据增强、是否使用位置编码等问题对模型进行更广泛的训练。研究表示,使用数据增强、位置编码以及超参数优化等方法均可提高模型准确率,稳定模型训练。评估结果汇总在表2中。结果表示,在扩增10倍的单反应物和双反应物数据集上,RetroTRAE在精确匹配(Tc = 1.0)方面达到了56.4%和60.1%的准确率。同时,作者适当放宽阈值提高模型成功率,当允许单片段突变(SM)时,单反应物和双反应物的成功率分别增加到58.1%和60.9%。允许双片段突变(DM)时,相应的预测结果提高为60.5%和62.7%。 表2 RetroTRAE模型预测准确率******[1]**********
2.模型的可解释性
作者通过观察注意力权重,以解释该模型实际上学到了什么。RetroTRAE模型更关注反应中心附近的AEs变化,例如开环反应等,如图2所示。这充分证明,与SMILES描述符相比,AE描述符是有化学意义的,而且本身是可以完全解释的。且该模型可与适当的搜索算法(如蒙特卡罗树搜索)相结合,预测多步逆合成路线。 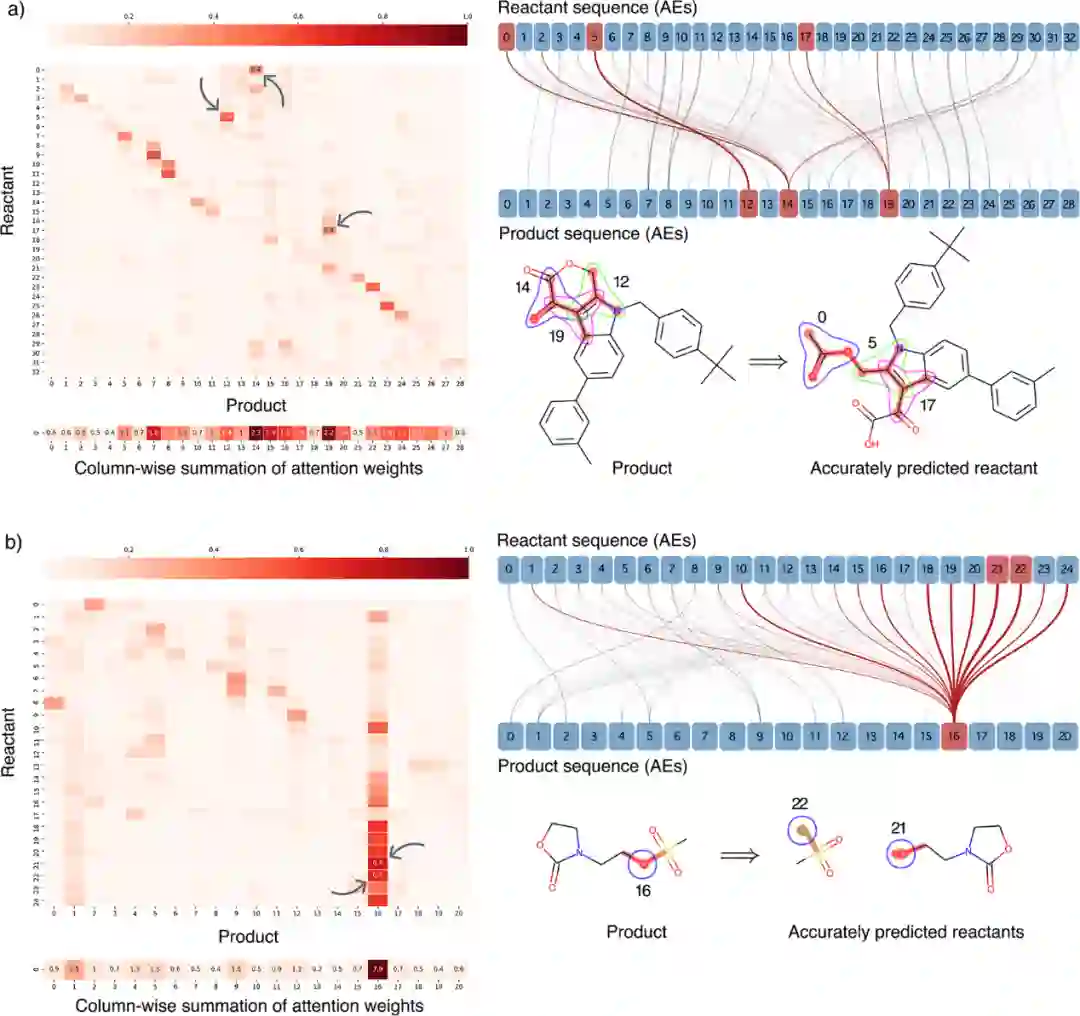
如图3所示,作者介绍针对硬阈值产生的三种预测结果,首先是为精确预测(图3a),RetroTRAE模型在测试集中的精确预测率为58.1%。其次是发生SM和DM的情况(图3b、图3c)。据统计,解码发生SM和DM的数量共占总预测量的3.3%。为了证明硬阈值(SM/DM)设定的可行性,作者随机选择了10个SM对和10个DM对,比较单片段和双片段突变与真实分子之间的相似性。研究表明,20对结构的平均Tc为0.91,RDKit产生的指纹图谱两两相似度为0.97,这些结果表明,硬阈值(SM/DM)所获得的预测结果是较为可靠的。 在单片段突变(SM)情况下,所有相连的原子类型都要与真实分子相同,因此只可能会发生有两种类型的结构变化。首先,由于单一原子环境的错位(例如,在邻位/间位/对位),可能出现一个新的原子环境(或现有的环境消失)。其次,在化合物末端增加或减少一个现有的AE。双片段突变(DM)的情况一般发生在错位的侧链AE或单原子的替换。
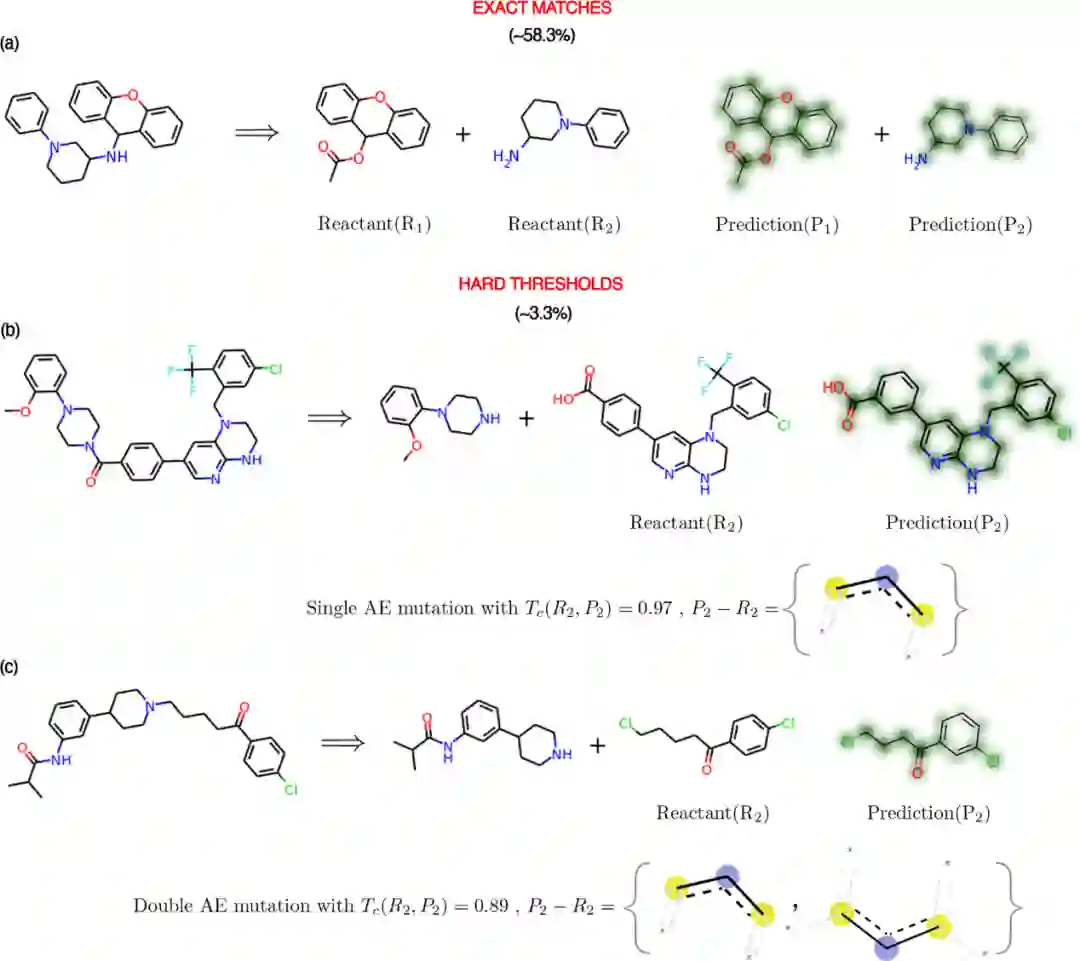
图3 RetroTRAE模型样例(a)精准预测(b)单片段突变(c)双片段突变,颜色表示原子级对总体相似度的贡献(绿色:相似度得分增加,红色:相似度得分减少,未着色:无影响)******[1]**********4.与现有模型比较
由于模型没有预先提供反应类信息,因此作者将该方法与其他不考虑反应类标记的逆合成预测方法进行了比较。结果如表3所示。RetroTRAE模型达到了58.3%的平均top-1准确率,超过了现有的基于NMT的无模板模型。允许SM和DM时,模型准确率提高到61.6%,是目前逆合成模型的最佳水平。表3 无反应类别的逆合成预测模型的top-1准确率比较******[1]**********

5.通过原子环境检索候选反应物
使用RetroTRAE模型进行预测后,得到的结果是预测反应物的AE的集合,可以通过数据库搜索来检索,成功检索到即证明模型预测的AEs可以完全还原为真实分子或高度相似的分子。作者使用PubChem研究了1000个USPTO测试分子检索反应物候选的成功率。检索测试结果显示,超过一半的预测(55.7%)可以被准确检索(图4)。允许SM后,检索成功率提高了约30%。当允许DM时,所有的测试分子都能被成功检索到。这证明模型得到的所有结果最多只有两个AE的差异。以上结果表明,用AEs表示和预测分子是一种可行实用的方法。
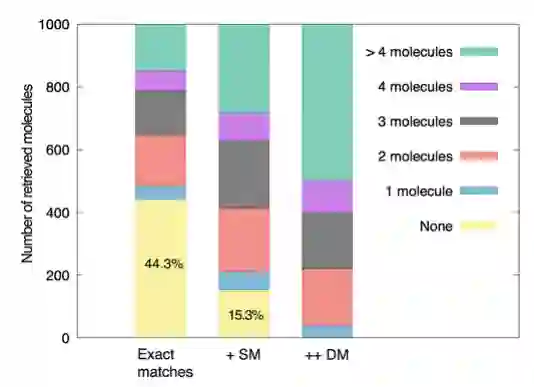
图4 在大型PubChem化合物库检索候选反应物****[1]********************总结
作者提出结合Transformer架构和原子环境(AE)表示法,开发了新的无模板逆合成预测模型,即RetroTRAE。实验证明,与传统的基于SMILES的逆合成预测模型相比,使用AE作为描述符进行逆合成预测精度提高,且具有可解释性,同时解决结构指纹在无模板的逆合成方法中的应用问题。该研究结果将为利用序列数据开发化学的NMT模型提供新的可能性,相信这种方法在有机化学中具有广阔的应用前景。 **参考文献 **
[1] Ucak UV, Ashyrmamatov I, Ko J, et al. Retrosynthetic reaction pathway prediction through neural machine translation of atomic environments. Nat Commun, 2022. 13(1): p. 1186.
供稿:张红文
校稿:刁妍妍/张梦婷编辑:毛丽韫华东理工大学/上海市新药设计重点实验室/李洪林教授课题组▼招聘博后▼华东理工大学李洪林教授团队诚聘博士后


Li's Lab地址:上海市梅陇路130号 电话:021-64250213课题组网站:http://www.lilab-ecust.cn 长按扫码可关注
