
作者 | 应俊杰 指导 | 闵小平(厦门大学)今天给大家介绍的是麻省理工学院林肯实验室、电子研究实验室等机构发表在arxiv上的预印文章《Antibody Representation Learning for Drug Discovery》。作者开发了一个抗体序列特定的预训练语言模型。发现在通用蛋白质序列数据集上对模型进行预训练可以更好的支持特征细化和学习抗体结合预测。证明语言模型能够在抗体结合预测方面学习到比传统抗体序列特征或CNN模型学习的特征都更有效的特征。分析了训练数据大小对预训练模型性能的敏感性。

研究背景
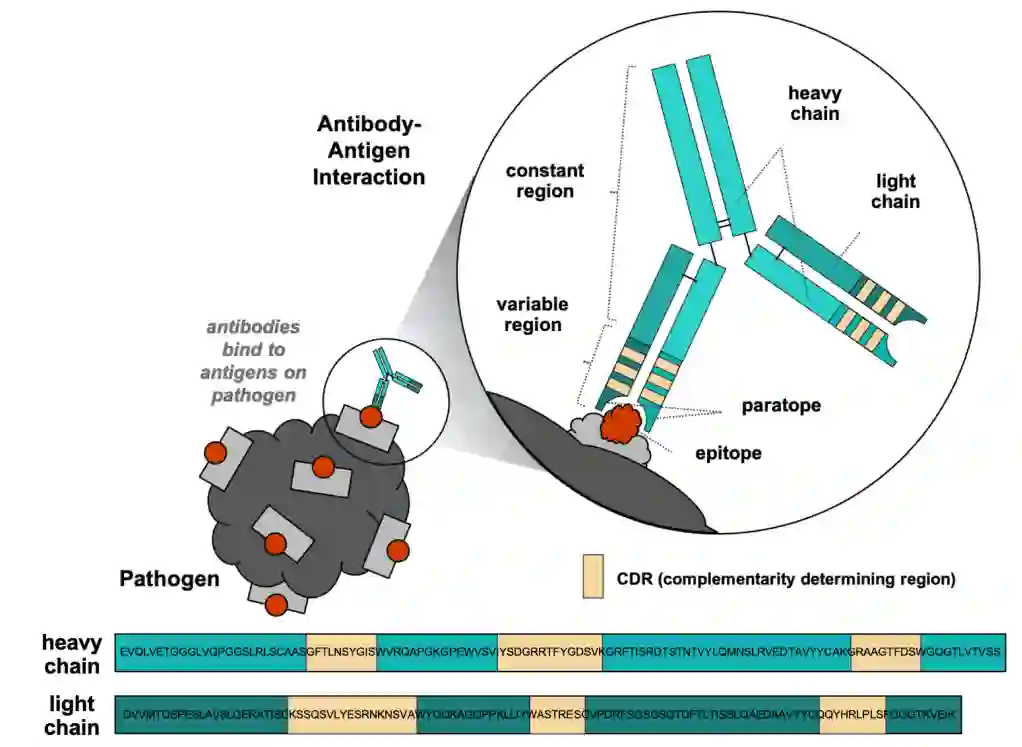
图1:抗体结构及其与抗原相互作用
抗体是由免疫系统产生的 Y 形蛋白质,用于标记或中和称为抗原的外来物质。如图1所示,抗体由两条相同的轻链和两条相同的重链组成。每条链都有一个可变区和一个恒定区。可变区的尖端形成抗体的结合表面,也称为互补位。该区域识别并结合特定抗原的结合表面称为表位。每条链的可变区包含互补决定区 (CDR),分别表示为 CDR-L1、CDR-L2、CDR-L3 和 CDRH1、CDR-H2、CDR-H3分别位于轻链和重链。CDR的氨基酸序列决定了抗体将结合的抗原。抗体设计主要就是设计其CDR区。在针对特定靶标设计优化的候选抗体时,会考虑抗体的许多天然特性,包括结合亲和力、结合特异性、稳定性、溶解度和效应器功能。抗体结合亲和力是本文重点讨论的特性。
抗体数据集
为了评估和比较学习的表示和传统的抗体特征,作者为抗体结合预测任务准备了以下几个基准数据集。
无标签的序列数据 Pfam:Pfam是知名的精选蛋白质家族数据库,其中包含单个蛋白质结构域的氨基酸原始序列。其中训练集、验证集和测试集分别包含 32,593,668、1,715,454 和 44311 个序列。
OSA(Observed Antibody Space):数据库是一个收集和注释免疫组库以用于大规模分析的项目。它包含来自超过 75 项研究的超过 10 亿个原始抗体序列。具体来说,轻链训练集、验证集和测试集分别包含 70,059,824、364,332和 414,635 个序列,重链数据训练集、验证集和测试集分别包含172,524,747 、46,603,347和51,043,837 个序列。
有标签的序列数据 LL-SARS-CoV-2 data:这是SARS-CoV-2 靶标(冠状病毒中的一种保守肽)的抗体序列的标记结合亲和力数据集(Walsh et al. 2021)。数据集是使用高通量方法生成的,用于计算机抗体库设计和抗体结合亲和力测量,数据集中亲和力基于AlphaSeq 技术测量。数据集中14L 为 25,474条,14H 为 25,210条,91H 为 21,577条,95L 为 29,384条。其中“H”表示重链, “L“ 表示所选抗体骨架的轻链。样本以 0.8/0.1/0.1 的比例随机分成训练/验证/测试集。
IgG抗体数据集:CDRH3 区域序列数据集。训练、验证和测试规模分别为 60,992、6,777 和 29,007。肽段长度可变,长度范围为 8 到 20。
特征表征
在本节中,作者总结了几类非 ML 和 ML 学习的抗体特征表示,以及它们如何用于训练 ML 模型进行预测。
传统特征表征 位置特异性评分矩阵(PSSM)是生物序列中最常用的表示之一。在多序列比对中给定一组 N 个功能相关的序列(固定序列长度 L),即 Sn = [xi, x2,····, xL] 和 n ∈ (1,····, N)
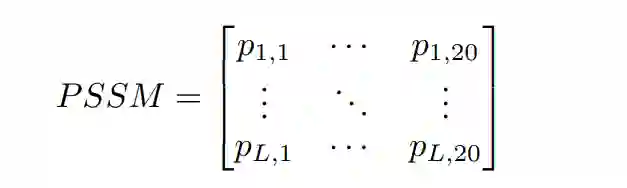
其中列代表20个标准氨基酸词汇[a1,a2,····,a20],pi,j表示序列第i个位置的氨基酸突变为氨基酸aj的概率。接着从PSSM中提取序列的向量表示,我们遵循 (Zahiri et al. 2013) 中提出的方法。也就是说,每个序列都由一个 400 个元素的向量表示。
有监督的表征学习 有了足够数量的标记数据,卷积神经网络 (CNN) 和长短期记忆网络 (LSTM) 已被用于训练抗体特征表示以进行亲和力预测。本文中,作者选择使用 CNN 作为监督表示学习的代表模型。
自监督语言模型 随着大量未标记的生物序列,自监督学习近年来在学习序列表示方面受到了极大的关注。预训练的语言模型包括 BERTTransformer 和扩张残差网络 (ResNet),它们使用掩码预测进行训练。自监督语言模型的性能主要取决于训练数据量、数据质量和语言模型的选择。为了了解训练数据如何影响语言模型的性能,作者用BERTTransformer 掩码语言模型来作为实验模型。
实验
预训练模型比较 为了了解训练数据如何影响语言模型的性能,作者训练了三个 BERTTransformer 模型(1)在 Pfam 数据集上训练的 Pfam 模型;(2)在OAS重链序列上训练的重链模型;(3)在OAS轻链序列上训练的轻链模型。首先根据各自的测试数据对语言模型进行评估。Pfam 模型、重链模型和轻链模型的平均困惑度分别为 13.1508、1.5990 和 1.4316。
为了在下游任务上比较这些预训练的语言模型,作者探索了两种迁移学习方法。一是在下游回归任务上微调预训练模型。二是冻结氨基酸嵌入层,然后构建一个序列长度不变的嵌入
表1:通过微调预训练语言模型预测抗体结合亲和力的 Pearson 相关性
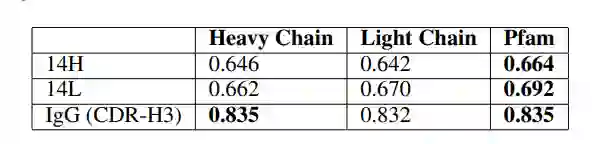
表2::通过在预训练语言模型中提取的特征训练高斯过程模型来预测抗体结合亲和力的Pearson 相关性
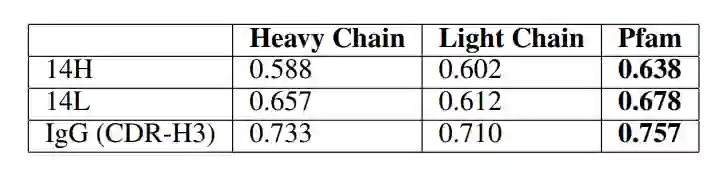
可以观察到,在不同的预测任务中,在更多样化的 Pfam 数据上训练的语言模型始终优于在抗体特异性轻链和重链数据上训练的模型。这表明了训练数据多样性的重要性。虽然蛋白质序列可能表现出不同的潜在分布,但从学习蛋白质语言模型中获得的知识可以捕捉到更高层次的生物学原理,这些原理可以转移和细化到抗体特定的任务中。对于接下来特征表示比较的实验,预训练的语言模型是都是在 Pfam 数据上训练的模型。
特征表示模型的比较 作者比较了三类特征表示:1.传统的基于 PSSM 的特征2.通过对标记数据进行监督学习产生的特征3.通过预训练的语言模型学习的特征。为了比较这些特征,作者用了不同的回归模型:MLP、岭回归和高斯过程 (GP) 回归。图 2 分别显示了预测性能。
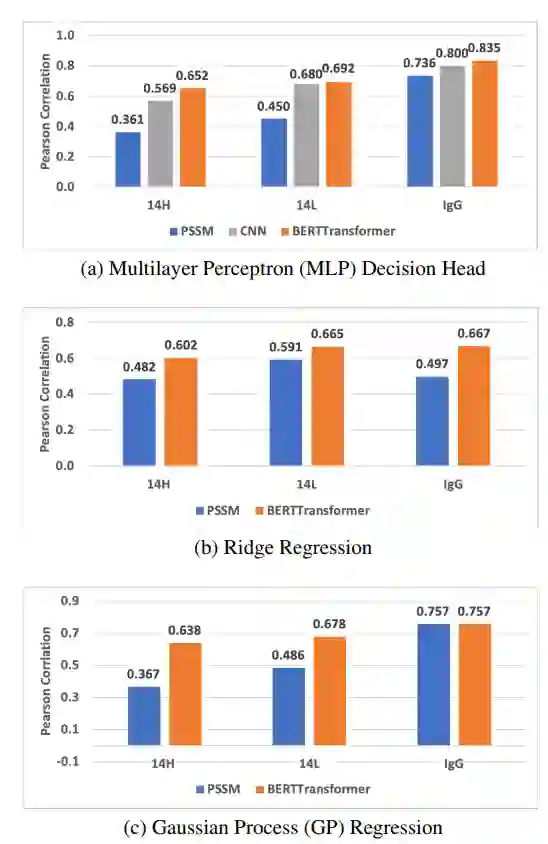
图2:14H、14L 和 IgG 亲和力预测任务的特征表示比较
总体而言,所有学习实验的结果表明,与其他特征学习方法相比,预训练模型能够更好地学习到抗体特征来进行结合预测。基于 PSSM 的特征的性能高度依赖于回归模型和训练任务。例如,岭回归模型在 14L 和 14H 任务上表现更好,而高斯过程在使用基于 PSSM 的特征的 IgG 任务上表现更好。
训练数据大小依赖性 作者研究了训练数据大小对抗体结合亲和力预测任务的模型性能的依赖性。使用不同的训练/验证子样本并平均 Pearson 相关性,从每个训练规模的五次独立运行中获得模型性能。对于每次运行,训练和验证数据都是随机子采样的,其中验证子样本是训练子样本的 10%。相同的随机训练/验证拆分用于训练比较的所有模型。所有模型都在完整的测试集上进行评估。图 3 显示了模型在 14L、14H 和 IgG 数据集上的性能。对于 14L 和 14H,岭回归是在基于 PSSM 的特征上训练的,而对于 IgG,使用高斯过程 (GP) 回归,因为 GP 在基于 PSSM 的特征上的表现要好于其他回归模型。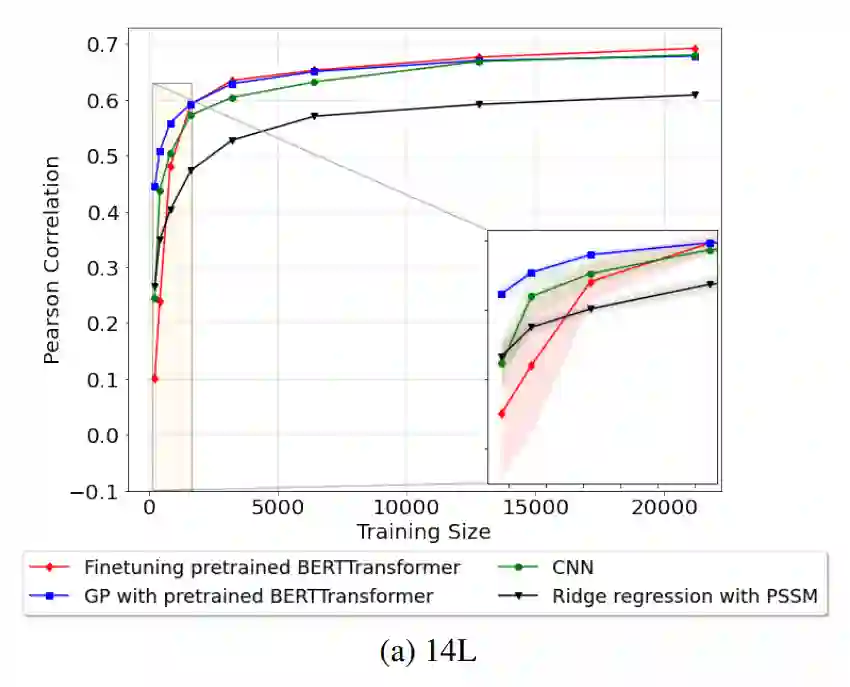
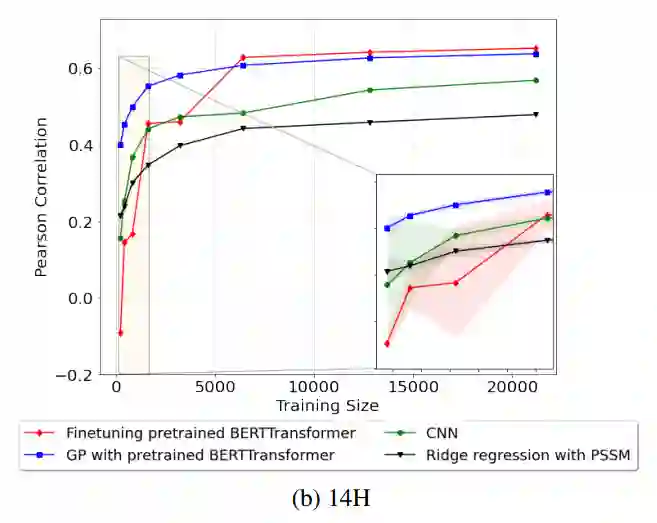
图3:14L、14H 和 IgG 的训练大小依赖性
总体而言,当训练数据充足时,微调预训练语言的性能始终优于其他模型。对于一小部分训练数据,在从预训练模型中提取的特征上训练的 GP 在不同的训练子样本上往往表现更好,性能差异更小,并且也优于传统方法,表明预训练模型的鲁棒性支持迁移学习的训练模型。3200 到 6400 之间的训练规模提供了最大幅度的性能改进,可用于指导样本量的选择。
总结
作者发现预训练中使用最多样化和通用的数据集 (Pfam) 时,亲和力性能最好。该结果支持抗体结构和功能特性不仅仅由抗体序列决定的假设。很可能还依赖于在一般蛋白质序列中更广泛遇到的其他特征。其次,作者发现大小在 3200 到 6400 个样本之间的训练集在亲和力绑定任务中提供了最高的性能增益。 参考资料 https://arxiv.org/abs/2210.02881
