编译 | 董靖鑫 审稿 | 张翔今天给大家介绍的是卡内基梅隆大学的研究团队近期的一篇分子优化工作。识别蛋白质抑制剂通常需要预测配体结合自由能(Binding Free Energy, BFE)。热力学积分(Thermodynamics integration , TI)是一种能够准确预测BFE的方法,但其计算成本高、时间长。**在这项工作中,作者利用自动机器学习(Automated machine learning, AutoML)和主动学习(Active Learning, AL)的技术开发了一个高效的自动化工作流程,只需要数百次TI计算能在数千个同源配体中识别BFE最低的化合物。**实验结果表明,该框架预测的化合物的结合亲和度显著提高。
1 介绍 药物设计的先导化合物的发现及优化阶段旨在发现先导化合物,即通过改变分子的化学结构来改善与生物靶点的结合亲和力,以证明其对靶点具有活性。因此,结合自由能的预测至关重要,而相对自由能(relative BFE, RBFE)指的是新配体与先导化合物之间的结合自由能差异。目前基于大规模分子库的虚拟筛选方法的性能因较差的预测配体排序的能力而受到限制。分子动力学模拟(Molecular Dynamics,MD)方法虽然可以较为准确地计算RBFE,但对计算资源需求高。为了有效地克服这些限制,作者提出了一个自动化框架计算RBFE,以优化先导化合物。
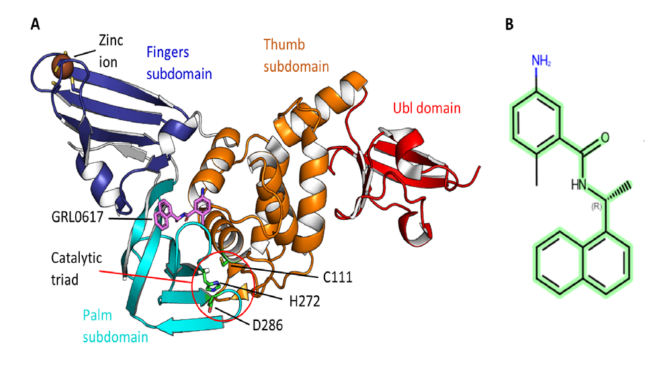
图1 SARS-CoV-2 PLpro及其抑制剂的结构
SARS-CoV-2木瓜样蛋白酶(papain-like protease, PLpro)是设计COVID-19抗病毒药物最有吸引力的药物靶点之一。研究表明GRL0617通用骨架(N-[(1R)-1-arylethyl]arenecarboxamide, 图1B)的结构对配体与PLpro结合很重要,并且在具有该骨架的化合物中可能发现更有效的PLpro抑制剂。在这项工作中,作者从13亿商业可用化合物库中筛选,选择了1万个N-[(1R)-1-arylethyl]arenecarboxamide衍生物的库,并最终确定了16种有效的结合剂,其预测的结合亲和力提高了100倍以上。
2 模型
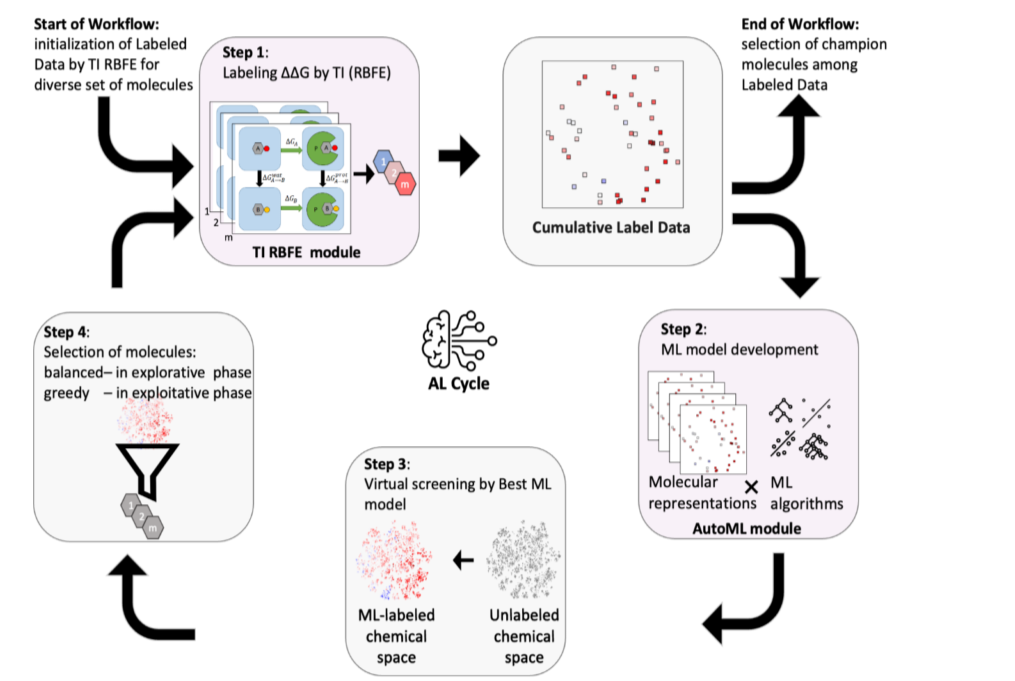
图2 模型流程
2.1 主动学习周期
两个主要的计算模块:
(1)AutoML模块。负责根据第二个计算模块提供的标记数据开发ML模型。 (2)TI RBFE模块。负责计算选定化合物与PLpro蛋白的相对结合自由能。
具体流程:
(1) 从分子的种子集开始,执行TI RBFE计算以获得训练数据和初始化的ML模型。 (2) 使用该模型筛选化学空间。 (3) 选择最佳候选分子集用于RBFE的TI MD计算。 (4) 对所选分子进行TI MD计算,并且用更新的数据重新训练ML模型。
2.2 自动化机器学习模块
使用基于先验选择的ML方法(如神经网络)和分子表征(如配体-蛋白质相互作用指纹)构建的ML模型可能会导致大量的模型偏差和样本选择偏差。AutoML旨在以数据驱动、客观和自动化的方式为ML模型选择、数据表示和超参数做出决策。AutoML和AL方法的结合既可以保证对化学空间进行快速、系统、公正的探索,又可选择最佳候选分子。
2.3 热力学积分分子动力学模拟模块
热力学积分的流程: (1) 生成MD输入文件(包括分子拓扑结构、原子初始坐标和约束)。 (2) 利用TI设置并行化的GPU加速的 MD模拟。 (3) 收集和处理输出数据。
3 实验 3.1 实验设置
AutoML-AL方法共进行8个AL周期。周期0中,使用多样的分子初始化AL,以尽可能广泛地采样化学空间。为初始分子集进行TI MD 计算,并提供给AutoML模块用于初始ML模型。周期1-5中,均匀地选择了具有低RBFE的分子,以获得库的化学空间信息。周期6,随机选择分子,这种方式有助于克服AL被困在化学空间的局部最小值时可能出现的问题。
3.2 实验结果
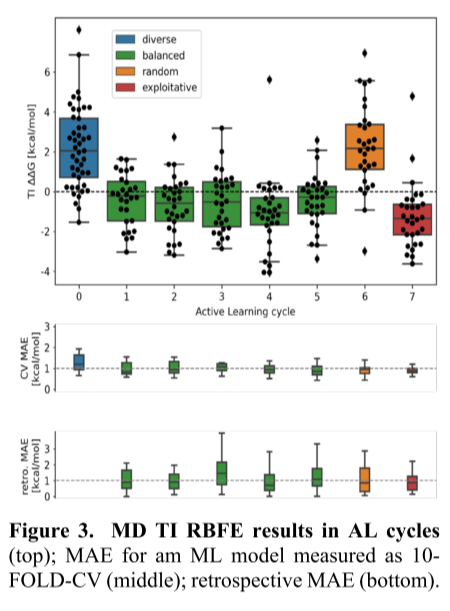
实验对253个配体进行TI MD RBFE计算。计算133种配体的RBFE为负的,约占TI计算的53%。这说明通过TI MD计算筛选的配体中有一半以上的配体具有比参考配体更高的结合亲和力。其中62种配体(占通过TI MD筛选的配体的24.5%)的预测结合亲和力提高了10倍以上。通过TI MD筛选得到的16个配体(占6%)与靶蛋白的预测结合亲和力提高了100倍以上。
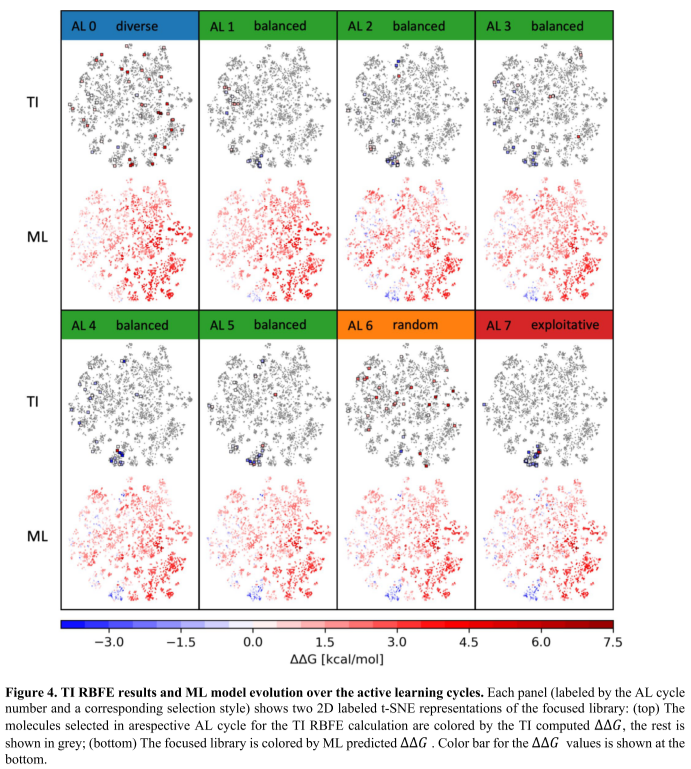
在AL周期0中,ML模型没有区分有利于结合的化学空间区域。在平衡选择的周期1-5中,模型探索多个区域,寻找期望的化学空间。由于信息的增加,ML模型的感知发生了明显的变化,开始识别出低∆∆G分子密集分布的化学空间区域。周期5结束,ML模型得到收敛,化学空间各个区域稳定着色。在随机选择分子的周期6中,分子分布在化学空间中,大多数分子如预期的那样具有正∆∆G。值得注意的是,模型的误差没有增加,这也说明了模型已经收敛。
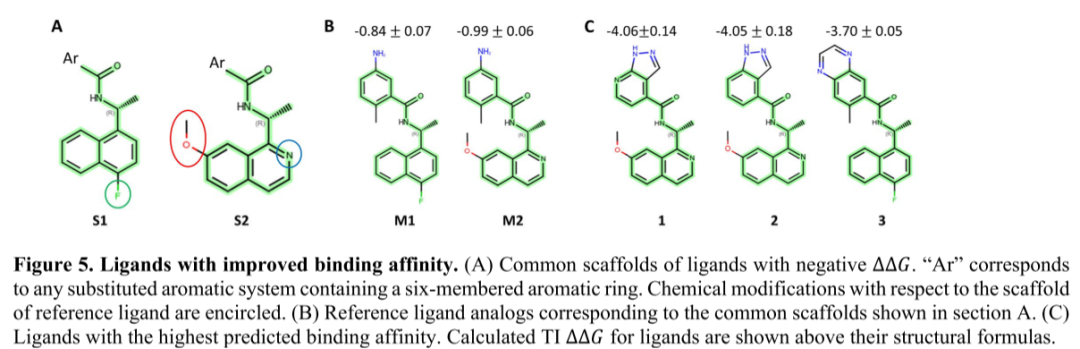
图5展示了两个优化的分子S1和S2,它们的结合亲和力分别提升了-0.84 kcal/mol和-0.99 kcal/mol。在TI ∆∆𝐺为负的配体中,有35个(~26%)分子具有相似的芳香体系。其中9个分子的预测结合亲和力提高了100倍以上。
4 总结 在这项工作中,作者提出结合自动机器学习(Automated machine learning, AutoML)和主动学习(Active Learning, AL)的方法对配体进行RBFE计算,实验表明该方法选择的配体结合亲和力显著提升。
参考资料 Gusev F, Gutkin E, Kurnikova M G, et al. Active learning guided drug design lead optimization based on relative binding free energy modeling[J]. 2022.