2022年10月28日,来自巴基斯坦的研究团队在期刊Journal of Biomolecular Structure and Dynamics上发表综述论文Deep learning in drug discovery: a futuristic modality to materialize the large datasets for cheminformatics。论文全面总结了人工智能方法在药物发现中的应用,并针对Covid-19的药物发现过程进行了详细的分析。

1 摘要
人工智能的发展模仿了人脑的工作原理来理解现代问题。传统的方法,如高通量筛选(high-throughput screening, HTS)和组合化学,由于只能处理较小的数据集,这对制药业来说是漫长而昂贵的。深度学习是一种精巧的人工智能方法,它可以对特定系统进行全面的理解。
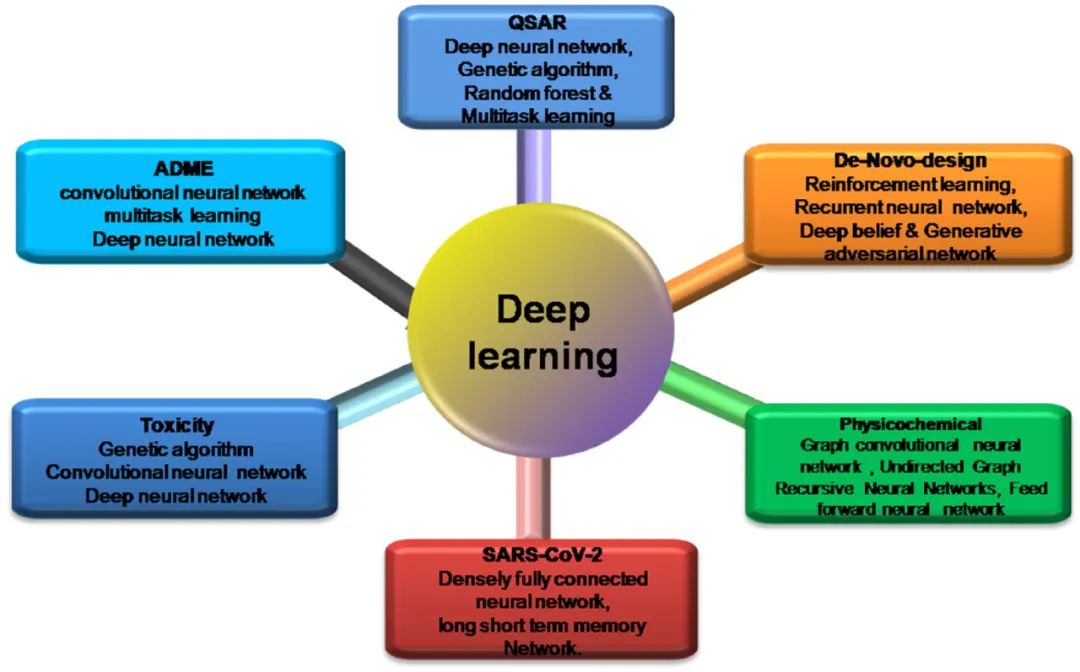
目前,制药业正在采用深度学习技术来加强研究和开发过程。多向算法在QSAR分析、药物从头设计、ADME评估、物理化学分析、临床前开发,以及临床试验数据精确性方面发挥着至关重要的作用。在这项研究中,作者调查了多种算法的性能,包括深度神经网络(deep neural networks, DNN)、卷积神经网络(convolutional neural networks, CNN)和多任务学习(multi-task learning, MTL)。研究表明,卷积神经网络、循环神经网络和深度信念网络(deep belief network)对于药效学属性的分子描述是兼容的、准确的和有效的。在Covid-19中,现有的药理化合物也已经使用深度学习模型进行了重新设计。在没有Covid-19疫苗的情况下,药物瑞德西韦(remdesivir)和药物奥司他韦(oseltamivir)已被广泛用于治疗严重的SARS-CoV-2感染。总之,这些结果表明了在药物发现过程中采用深度学习策略的潜在好处。
2 介绍
20世纪末,化学信息学被引入药物研发过程。湿实验方法开发药物成功率低、耗时长、花费高,药物研发过程转向了新的范式。通常,生成精确的先导化合物,且具有良好的吸收、分布、代谢、排泄、毒性(ADMET)性质和物理化学性质是该行业面临的最大挑战。计算工具有提高药物开发过程的分析准确性的潜力。计算机辅助基于结构的药物设计(structure-based drug design, SBDD)和基于配体的药物设计(ligand-based drug design, LBDD)方法在计算药物设计过程中占有一席之地。
多年以来,机器学习方法被用于不同的领域,但存在冗余和过拟合等问题。这些问题阻碍了机器学习模型的应用。2006年,Hinton等人设计了一个深度信念网络(deep belief network, DBN)。DBN由一组非线性隐藏层组成。它有助于克服冗余和过拟合问题。在深度学习中,Frank Rosenblatt于1957年首次创建了神经网络模式,并将其称为深度神经网络(deep neural network, DNN)。神经网络如今可以处理以前不可能的图像识别问题。监督学习、无监督学习和半监督学习是使用基于梯度下降的反向传播策略来克服冗余和过拟合问题的常用工具。
2.1 医疗中的深度学习
人工智能技术可以应用于医疗领域的很多方面(如图1所示),并被认为有助于医生的临床诊断。在心脏病学、糖尿病、视网膜病变、抗生素耐药基因和阿尔茨海默病研究中,各种深度学习模型被开发用于预测疾病状况和药物的疗效。已有研究模型可以精确地将心肌病分为限制性和收缩性病理。除了心血管、代谢和其他疾病,人工智能还可以改善肿瘤诊断。深度学习模型有效地指示了新冠肺炎模式,并对疾病进行了分类。
2.2 药物设计中的深度学习
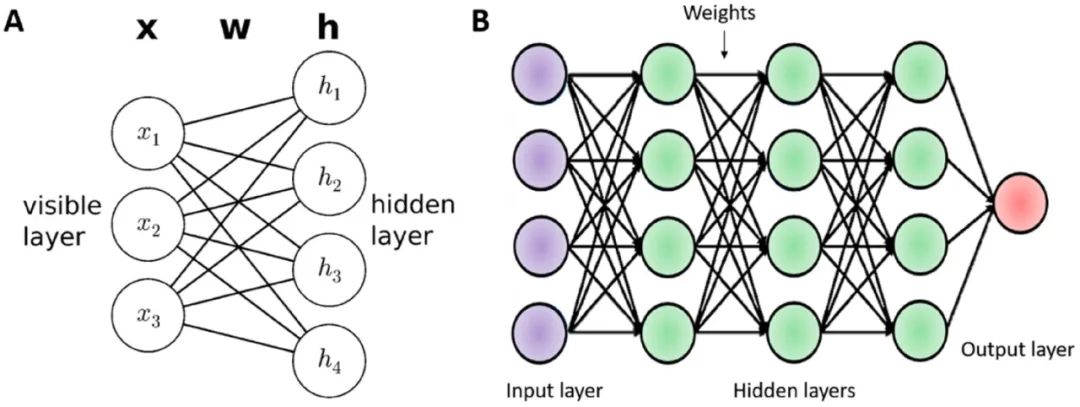
深度学习是机器学习的一个子类,更广泛地说是人工智能的一个子类。在机器学习中,新特征的处理和生成通常基于小数据集,通过支持向量机(SVM)、随机森林(RF)、k最近邻(KNN)、线性回归、逻辑回归、朴素贝叶斯、决策树等算法进行处理,这些算法仅在二分类中进行。人工神经网络(artificial neural network, ANN)是人工智能的最简单形式,如图2所示。ANN由2-3层组成,包括单个输入层和隐藏层,然后是作为神经元的单个输出层。深度学习将大脑功能模拟为ANN。它包含一组多个隐藏层以提取所需信息。深度学习是一个自动化过程,使用图形处理单元(GPU)和张量处理单元(TPU)加速来确保计算任务的自动化。为了执行专门的任务,深度学习中的开源编程语言包括torch、Deeplearning4j、CNTK、python和R。深度学习可分为监督学习、无监督学习和强化学习。卷积神经网络(CNN)已经应用于自然语言处理(NLP)、图文摘要、图像分类和语音识别。CNN由三个基本层组成,称为卷积层、池化层和全连接层。CNN通过稀疏连接和共享权重融入卷积层提取特征。池化层完善了鲁棒性特征,并通过全连接层生成输出数据。另一方面,DNN架构是一种前馈算法,可以从输入数据中提取复杂特征,并将其映射到输出模式中。DNN有多个隐藏层网络,可以自动处理输入数据。深度生成算法(deep generative algorithm, DGA)模型是一种基于无监督学习结构的模型,可以从高维输入数据源中提取未标记的数据。深度信念网络(DBN)和受限玻尔兹曼机器(RBM)是类似模型的例子。循环神经网络(RNN)不仅基于输入数据,还基于先前事件的输入序列。RNN通过将单个元素保存在在隐藏层单元中并连接相邻的隐藏节点来为序列数据处理提供预测。RNN使用反向传播训练以最小化模型中的误差。长短期记忆神经网络(LSTM)是RNN另一种变体。这都是药物设计和开发过程中最常见的算法。
3 QSAR预测
Neves等人2018年开发一种经济、快速且高效的工具建模定量结构-活性关系(quantitative structure–activity relationship, QSAR)。该方法广泛应用于预测定量结构-活性关系,其基于化学结构的描述性统计。高通量筛选通过为精确筛选提供丰富的数据,提高了QSAR的有效性。由于提取新的苗头化合物的高成本,HTS的使用受到限制。QSAR存在活性悬崖问题。虽然对特定蛋白质靶标具有活性,但结构相似的化合物在抑制潜力方面表现出差异,并改变了QSAR反应。出现这个问题的原因可能是工具选择了效率低的描述符,也有可能是学习只是在浅层中进行。深度学习模型在QSAR评估过程中缓解了此类问题。深度神经网络模型中高效开发的算法有可能克服活性悬崖、不准确和其他与QSAR方法有关的问题。因此,深度学习模型广泛应用于QSAR预测,如AtomNet。它是一种基于深度卷积神经网络的方法,可以从一个独特的层面预测具有更好结合模式的新化合物。它针对在57%以上的靶标,对于AUC指标显示出0.9(满分1)的精度,这高于已知的对接模型。其优于先前使用的RF和SVM模型,证明了深度学习模型在确定化学结构生物活性特征方面的能力。
4 药物从头设计
药物从头设计是一种基于特定靶标所需特征构建新化学实体的计算技术。深度学习模型,改进了从头开始的过程。一些算法提高这些模型的效率,如强化学习在碳和氮原子位置交换、优化分子性质和手性化合物形成方面具有优势,可以提高发现苗头化合物的效率。2019年,Niclas Stahl等人使用强化学习生成了2,048,000个化学结构,其中387个化合物含有所需的理想性质和分子特征。该模型基于365,521个化学结构进行训练。较大的数据集增强了强化学习在选择最佳分子特征方面的学习能力。该模型表明,大数据集提高了模型设计发现具有良好特征的新化合物的效率。
除了基于RL的模型,基于RNN的模型也被用于药物从头设计。有研究者将ChEMBL数据库中的50,000,000个SMILES格式的化合物输入RNN模型中开发新分子。该技术生成的864,880个化合物与输入数据和重复数据没有任何相似性。基于RNN的深度生成方法也可利用SMILES进行药物从头合成,如基于生成对抗网络(generative adversarial networks, GANs)和变分自动编码器(variational autoencoders, VAEs)。另外,多任务学习可以同时筛选合成化合物的多个化学性质,已有研究者将其应用于药物从头设计中。
5 ADME属性
吸收(Absorption)、分布(Distribution)、代谢(metabolism)和排泄(excretion),是被认为是药物开发过程的重要评估参数的药代动力学属性(见图3)。英国GlaxoSmithKline公司开创了成功的计算ADME建模。多任务学习被认为是药物开发过程中的一种有优势的模式。有研究者开发了多任务的图神经网络模型、多种机器学习工具结合方法用于预测化合物的ADME属性。
6 毒性预测
化合物在表现出治疗效果的同时,也可能对细胞体具有毒性作用。2014年,美国国家卫生研究院针对12,000种化合物的计算模型发起了Tox21挑战。其选择了650种化合物进行验证,主要针对与毒性问题高度相关的核受体信号通路(nuclear receptor signaling pathway, NR)和应激反应(stress response pathway, SR)通路。深度学习在机器学习和高通量筛选技术中表现最佳。随后,一场使用深度学习的比赛被举办。化合物中的毒物基团识别是筛选性质的主要任务,如电子供体/受体、芳香环或疏水区。在比赛中,有研究者使用扩展连接性指纹-4(extended connectivity fingerprint-4, ECFP4)进行化合物特征的二进制表示。多任务学习方法学习到了Tox21挑战数据中的独特特征,并从模型学习到细小特征(如磺酰基和毒性基团簇),接着提取了层次化的抽象特征。该模型在Tox21数据挑战的所有工具中处于领先地位,成功完成了15项任务中的8项。在Tox-21挑战之后,研究者开始了更广泛的探索,以发现更小的毒性结构。另外,也有研究者结合随机森林模型和DNN模型来提取毒性基团的特征。也有研究将遗传算法融入KNN分类模型中以选择相关描述符,来计算分子描述符的毒性分布。
7 物理化学性质
药物动力学参数的准确评估和预测对药物发现和开发过程有积极作用。深度学习模型在预测化合物的物理化学参数有着诸多应用。如,Moonshik Shin等人使用FDA推荐的人类结直肠癌细胞系(Caco-2)的深度神经网络模型来确定化合物的渗透性和药物转运性质。他们使用了两名独立研究人员提供的SMILES字符串。663种化合物用于训练和验证深度学习模型,剩余的用于测试模型的准确性;其还与目前使用的计算模型进行了比较,主要有:基于隐含狄利克雷分配(latent dirichlet allocation, LDA)的分类器和基于梯度增强(gradient boost, GBT)的分类器。深度神经网络模型取得了不错的表现。 在水溶性、亲脂性、电离常数pKa等性质的预测中,研究者也提出了诸多深度学习模型,例如深度学习模型在pka值的预测中产生了显著的结果。由于分子中存在多个离解/电离位点,这种预测对于其他模型都不方便。一项研究引入了人工神经网络模型,其基于大量数据集训练,并用少量数据集进行测试模型的准确性,发现该模型优于pka分析的传统计算方法。
药物-靶标结合是筛选化合物的基本过程。药物-靶标结合基于结构起作用,药物需要与靶蛋白的3D结构进行相互作用,而基于配体的结合通过已知抑制化合物的实验数据处理相互作用模式。原子卷积神经网络(Atomic Convolutional Neural Network, ACNN)是最新开发的基于原子CNN的工具,它分层提取空间特征。对于深度学习在物理化学性质预测方面的应用可以详见图4。
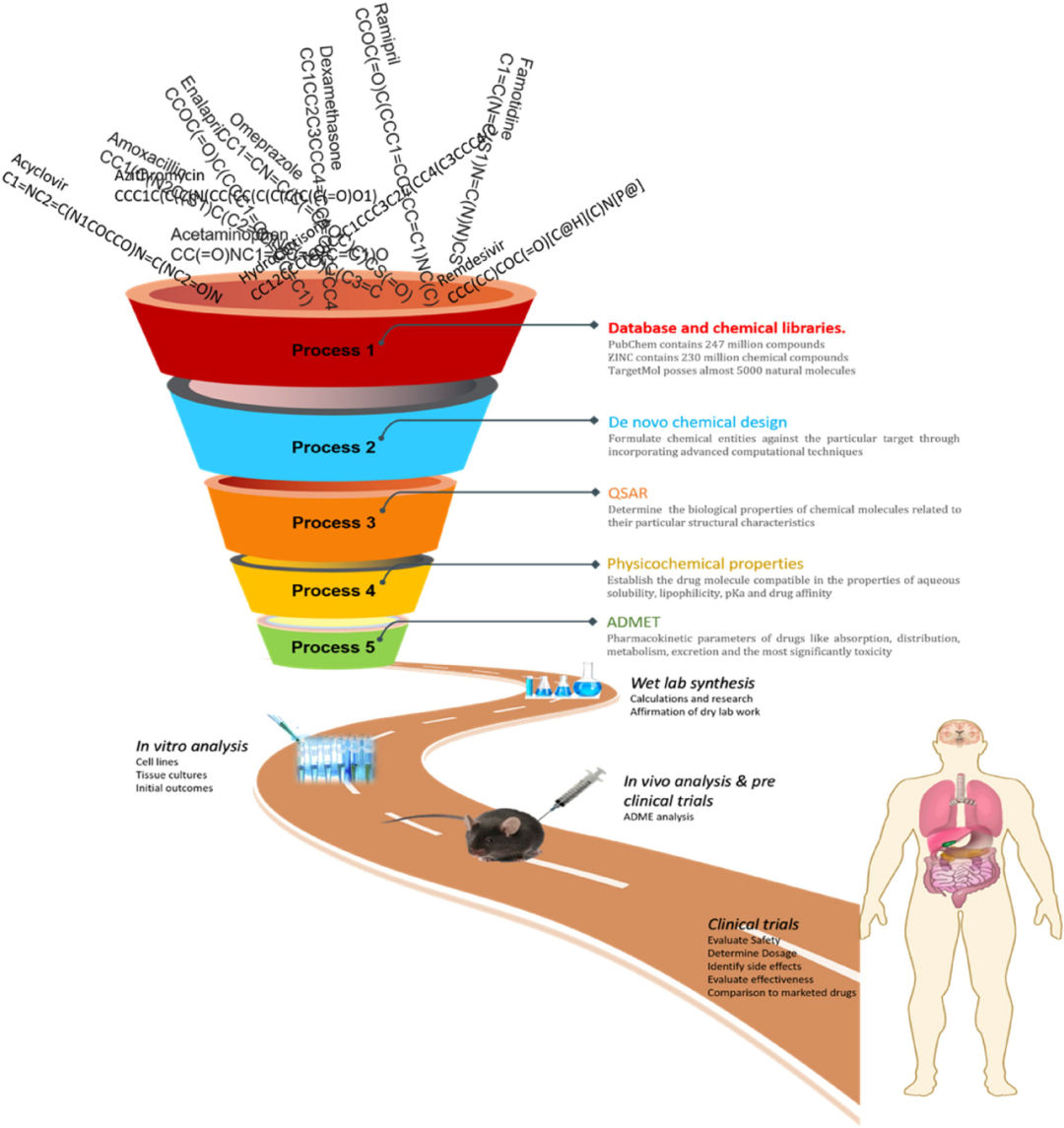
8 深度学习对防治传染病的意义
传染病是由细菌、病毒和真菌等微生物引起的。随着数学工具的引入,研究人员现在能够更好地预测流行病,理解每种传染病的特征,并确定可能的治疗靶标。本文回顾了一些算法,以证明深度学习在推动传染病抑制剂研究的应用。作者重点介绍了新冠肺炎抑制剂的相关研究,其中自然语言中的注意力机制也被引入其中,建立了基于卷积神经网络模型以研究药物-靶标相互作用(见图5),模型基于大量的化合物库进行训练,部分数据库如表1所示。
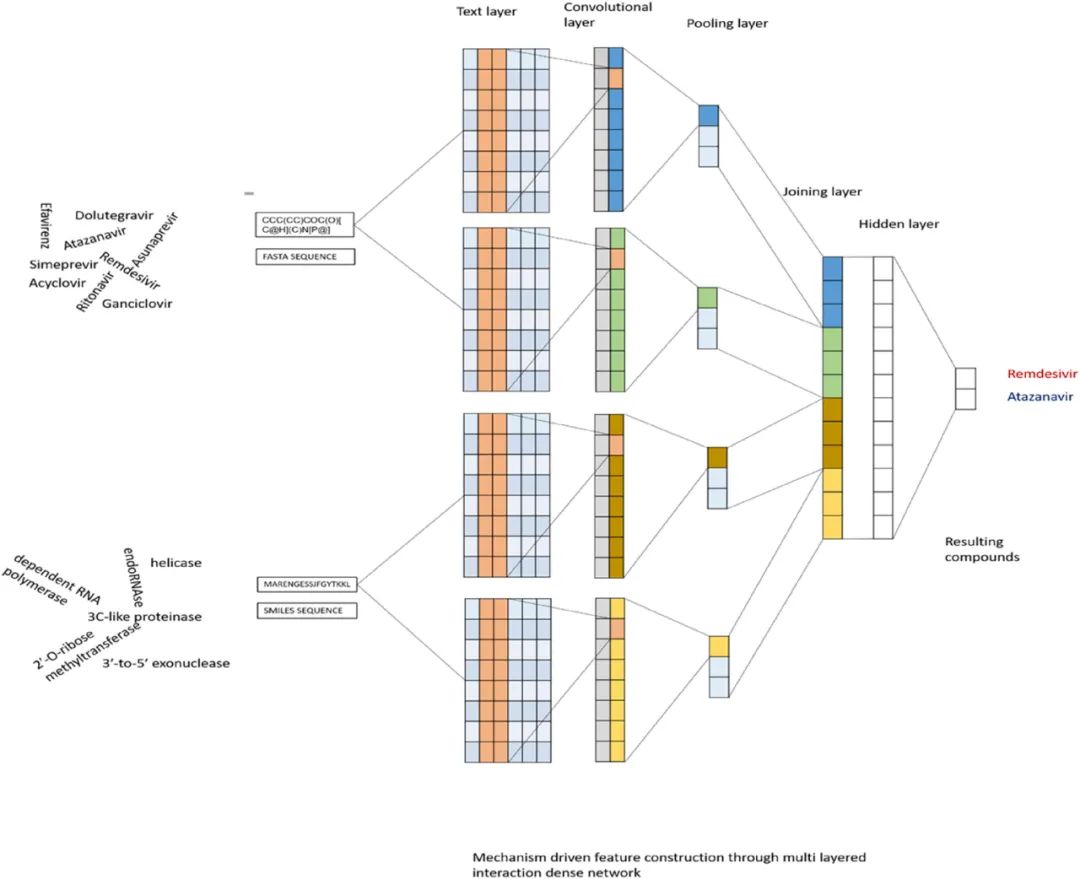
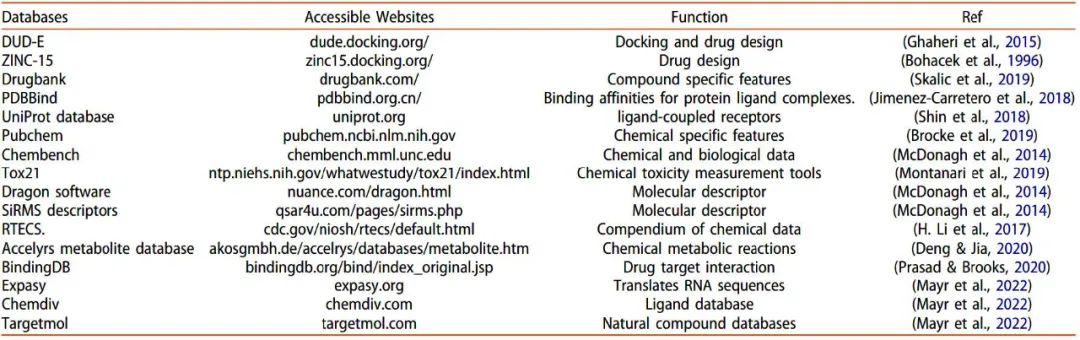
表1 可用于现代计算技术和深度学习方法化学/药物分子的化合物数据库
9 结论
人工智能正成为人类发展的基本组成部分,深度学习正通过逐步和稳定的潜力发展到科学领域,以推进开发可用的计算工具。新兴的深度学习工具对当前技术领域中可用于制药的方法提出了挑战。这些工具的目标是从现有的化学空间从头设计和开发化合物。论文介绍的数据库是基础化学数据的丰富来源。对于QSAR方法中的问题,如活性悬崖形成盐桥(activity cliffs forms salt bridge),AtomNet为靶标预测了更好的结合亲和力的药物。除了AtomNet,遗传算法和其他一些工具也有助于预测配体和受体的对接分数。循环神经网络、强化学习、深度信念网络和生成对抗网络等技术已被用于从头化合物生成,并报告了更好的H-供体(H-donors)、H-受体(H-acceptors)、可旋转键(rotatable bonds)、logP和总极性表面积(total polar surface area),这对于化学物作为候选药物的适用性至关重要。深度学习预测和开发具有这些性质的化合物的性能似乎与现有工具相当。药物研发的另一个最重要的性质是其化合物的ADME性质。多任务学习和图神经网络已被应用于预测这些性质。这些模型在识别实质性候选化合物及其改善作用方面发挥了潜在作用,这些化合物有成为成功的药物分子的潜力。这些方法显示的另一个重要方面是预测物理化学性质的可靠性。图神经网络、基于隐含狄利克雷分配的模型、深度神经网络等一些算法已经成功地用于预测水溶性、亲脂性和pka的活性。深度学习方法最可靠的服务是其应用在SARS-CoV-2大流行中的最新方法,该方法影响了世界各地的重要人群。这些方法优于传统方法,并发现了几种针对新冠肺炎靶标的新型药物化合物。尽管与传统技术相比,计算方法的结果稍好一些,但其局限性迫使研究者试图寻找潜在候选方案。因为它在某些方面被认为是一个黑盒子,但呈现的结果正在变得可靠。人工智能技术的成果正在浮出水面。参考资料Raza A, Chohan T A, Buabeid M, et al. Deep learning in drug discovery: a futuristic modality to materialize the large datasets for cheminformatics[J]. Journal of Biomolecular Structure and Dynamics, 2022: 1-16.