本文介绍的是由中南大学计算机科学与工程系的Lei Deng,Hui Wu等人发表在international journal of molecular science上的文章《DeepD2V: A Novel Deep Learning-Based Framework for Predicting Transcription Factor Binding Sites from Combined DNA Sequence》。转录因子(TF)是一种通过与位于启动子和增强子区域的上游调控元件结合来控制基因活性的蛋白质。本文提出了一种用于预测蛋白质- DNA结合位点的新型深度神经网络模型DeepD2V。 1.摘要预测人体内蛋白质-DNA结合位点在药物设计和开发等多个领域是一项具有挑战性和紧迫性的任务。大多数启动子包含大量转录因子(TF)结合位点,但目前仅有一小部分已通过实验鉴定出来。为了应对这一挑战,人们提出了许多计算方法来从DNA序列中预测TF结合位点。虽然以往的方法在预测蛋白质-DNA相互作用方面取得了显著的效果,但仍有相当大的改进空间。本文提出了一个混合深度学习框架,称为DeepD2V,用于转录因子结合位点的预测。首先,本文用一个原始DNA序列和它的三种变体序列,包括它的逆序列、互补序列和互补逆序列,构建输入矩阵:一个具有特定步幅的大小为k的滑动窗口用于获得输入序列的k-mer表示。接下来,本文使用word2vec获得预先训练的k-mer词分布式表示模型。最后,利用循环神经网络和卷积神经网络预测蛋白质与dna结合的概率。在50个公共ChIP-seq基准数据集上的实验结果表明,DeepD2V具有良好的性能和鲁棒性。此外,本文章中验证了使用基于word2vec的k-mer分布式表示的DeepD2V的性能优于单热编码。且卷积神经网络(CNN)和双向LSTM (bi-LSTM)的集成框架优于单独使用CNN或双LSTM模型。 2.数据集本文从ENCODE项目中收集了50个公开的ChIP-seq数据集,以评估所提出的DeepD2V方法的性能。数据集来自三种类型的细胞系,包括Gm12878, H1-hESC和K562。对于每个细胞系,从峰值文件中的每个记录中选择了约15000个顶级序列作为阳性样本,其中每个序列包含200个碱基对。同时生成负序列,以匹配正集的统计特性;否则,生成的数据集可能导致实验结果有偏差。阴性样本是根据Ghandis等人的工作,通过匹配阳性样本的重复分数、长度和GC含量生成的。本文章中生成了三个具有不同阴性与阳性样本比率的数据集,以评估文章所提方法的鲁棒性。负极与正极之比分别为1:1、2:1和3:1。为了评估DeepD2V的性能,文章采用了三重交叉验证来优化模型参数。将基准数据集随机分为三组,其中两组作为训练集,其余两组作为测试集。此过程重复了三次,计算了性能指标的平均值。此外,1/8训练样本随机抽样作为验证集。 3**.**** 序列转换与表示**
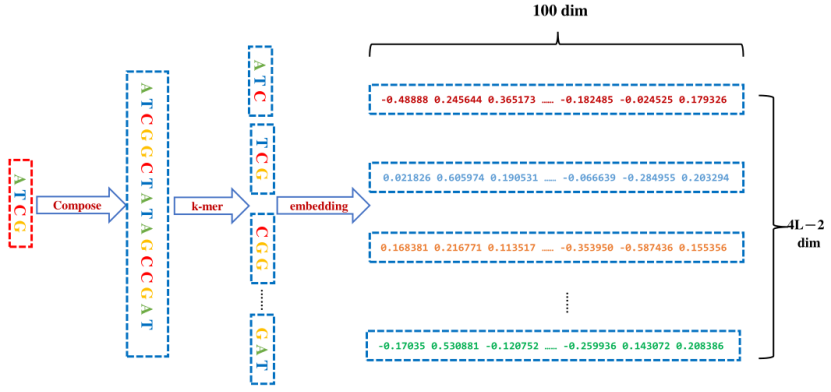
图1.序列转换表示图首先,从一个原始DNA序列推导出另外三个变异序列,包括互补序列、逆序列和互补逆序列。将四个序列按特定顺序连接到dna2vec方法中,得到分布式表示矩阵。然后,根据序列分析中常用的k-mer方法对组合序列进行分割。k -mer用于将一个序列分成多个以k为基数的子串。当步幅为1时,l个碱基的序列被分为(l−k + 1)个k-mer。例如,序列AGCCT被分成三个3-mers:(AGC, GCC, CCT)。因此,长度为l的DNA序列被拆分为具有k个碱基的k-mer子序列,形成一个长度为(l−k)/s+1的单词总数。本文把整个DNA序列看作一个句子,把k-mer片段看作组成句子的单词。因此,使用连续词袋模型(CBOW)模型来训练所有经过处理的序列语料库生成序列词向量。CBOW模型根据上下文预测目标词出现的概率。每个目标词都由一个100维向量表示。经过几轮迭代之后,每个长度为200 bps的原始基序列被转换为(798,100)矩阵。序列转换和表示如图1所示。 4.** 模型架构**

图2.网络架构本文中DeepD2V的图解如图2所示。卷积模块用于从输入序列中提取特征,Bi-LSTM模块用于捕获高阶特征,然后是两个全连接层和一个dropout层进行预测。与RNN相比,一维卷积神经网络可以缩短序列,提取基序的高阶特征,且处理较长的生物序列时需要较少的计算成本。因此,本文在RNN之前使用一维卷积模块。每个卷积模块由一维卷积层、ReLU层和最大池化层组成。卷积层负责用指定数量的滤波器内核捕获motif特征。采用Relu作为激活函数,可以有效地减少梯度下降和反向传播,避免梯度爆炸和梯度消失。最后,采用最大池化层来减少卷积层输出的冗余信息,并在滤波器中选择最大响应。长短时记忆(LSTM)是RNN的一种变体,解决了传统RNN不能长期依赖的问题。考虑到DNA序列的双链结构,选择bi-LSTM进行长期提取序列中的特征,t位置双LSTM单元具体计算公式如下图3所示:
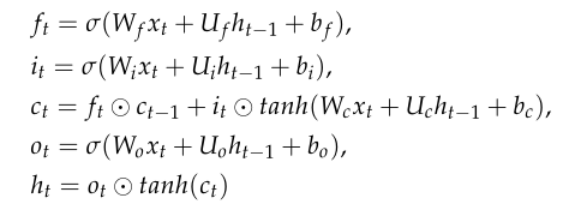
图3.LSTM具体计算公式上图中bo, bc, bi和bf是偏差,Wi, Wf, Wo, Uo, Uf和Ui是权重。两个全连通层和一个dropout层构成最后的预测模块,整合了从CNN和RNN模块中学习到的特征。Dropout被广泛应用于深度神经网络中,通过减少神经元之间复杂的自适应关系来进行正则化以避免过拟合。Dropout的比率通过交叉验证进行调优。最后,利用sigmoid函数计算蛋白质与dna结合的概率。 DeepD2V模型由PyTorch 1.0实现,源代码和数据集可在https://github.com/Sparkleiii/DeepD2V上获得。超参数从每个ChIP-seq数据集的搜索空间中随机采样,并通过交叉验证调整到最优值。本文通过Xavier均匀分布初始化所有权重,并初始化所有偏差为零。DeepD2V模型在20个周期内达到收敛,训练损失曲线逐渐减小,20个周期后趋于稳定。因此,本文将训练周期设置为20,选择验证集上ROC AUC值最高的模型作为最终的最佳模型。记录测试集上最佳模型的性能指标,并将三次交叉验证结果的平均值作为最终结果。根据前面研究的结果,将卷积核的个数设为16。 5. 实验结果本文首先用不同的步长和k-mer值评估了DeepD2V的鲁棒性。为了验证组合DNA序列对性能的改善,比较了不同输入条件下DeepD2V模型的性能。最后,将DeepD2V与仅使用CNN、RNN或Bi-LSTM模块的其他简化模型进行比较。为了进行系统性能评估,本文章将DeepD2V与其他四种最先进的蛋白质-DNA结合方法进行了比较,包括DanQ、DeepBind、WSCNN和WSCNNLSTM。在50个ChIP-seq基准数据集上进行了性能比较实验。鉴于本文使用dna2vec方法来获得每个k-mer的分布式矢量表示,通过实验比较k和s值的不同组合在Gm12878细胞系的23个ChIP-seq数据集上的性能,可以确定k和步长s的最佳值。如图4所示,当k的值增大时,模型的性能逐渐下降。该模型在k = 3时的性能更好。文章认为,这一结果可能是由于三个碱基组成一个氨基酸,而k = 3恰好抓住了序列的生物学本质。对于后续的步长s调整,于是将k设置为3,然后使用网格搜索s的最优值。
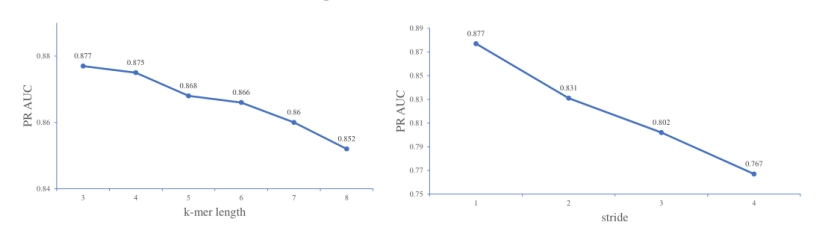
图4.不同k-mer长度和步长的比较图4显示,步幅越大,模型的性能越差。这一结果可能是由于大跨步导致了细粒度序列信息的丢失。先前的研究鼓励使用较大的k-mer长度值,并表明较小的步幅值(s = 1)可能会降低嵌入算法的性能。然而,在本文章的实证实验中,发现当stride和k-mer长度(k = 3和s = 1)值较小时,模型的性能优于其他k和s的组合。因此,在接下来的性能比较实验中,将k和s的值分别设为3和1。注意,该实验使用原始DNA序列作为输入,而不是组合序列。本文的模型采用word2vec来构造k-mers的分布式表示,而不是k-mers的单一热编码。虽然词嵌入技术已广泛应用于自然语言处理领域,但在蛋白质与dna结合的研究中仍很少应用。为了验证dna2vec的优势,本文在50个公开的体内ChIP-seq基准数据集上进行了几个比较实验。图5展示了使用单热编码和dna2vec的DeepD2V在平均AUC和f1评分指标下的性能。由此文章可以得出结论,使用dna2vec的DeepD2V在所有三个指标上都明显优于单热编码。
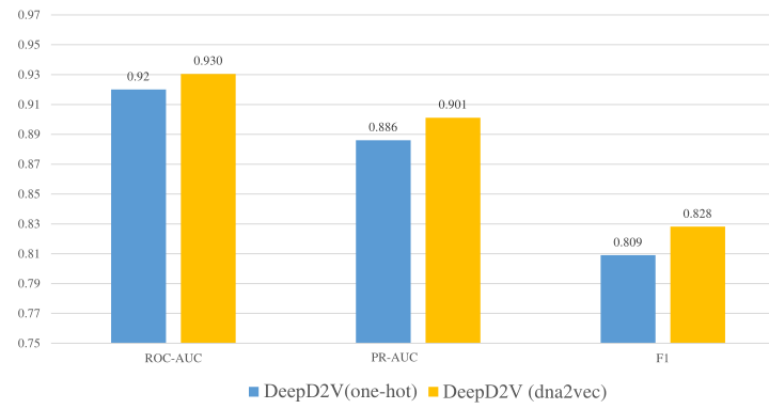
图5.使用单热编码的DeepD2V与dna2vec在50个活体数据集上的性能比较对于双链DNA序列,DeepD2V训练时会考虑反向或互补序列,因为蛋白质可能会与DNA互补链或反向序列链结合。因此,本文再次探讨了组合序列和原始DNA序列的意义。图6显示了DeepD2V在所有50个基准数据集上以原始DNA序列和组合序列作为输入的性能。可以发现,与原始DNA序列相比,组合序列在三个性能指标上都大大提高了DeepD2V的性能。认为组合序列可能捕获不同DNA序列模式之间的高阶依赖关系,如隐式DNA形状。
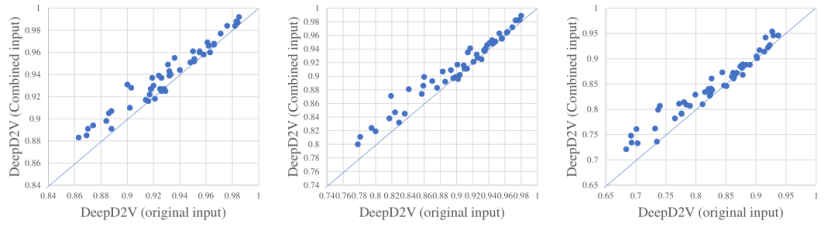
图6.原始输入和联合输入的比较为了验证结合CNN和bi-LSTM的性能改善,本文将DeepD2V与三种简化模型进行了比较:仅CNN、仅RNN以及CNN和RNN的混合结构。图7显示了使用这些深度学习架构的DeepD2V在所有50个公共ChIP-seq数据集上的性能。需要注意的是,对四个模型的具体超参数进行了优化,以便进行客观的性能评价。认为CNN能够很好地从序列中提取高阶特征,而bi-LSTM能够捕捉到基元之间的长期依赖性,避免了DNA长序列训练过程中梯度消失和梯度爆发的问题。与单独使用CNN或bi-LSTM相比,DeepD2V的预测性能更好,f1平均得分为0.809。DeepD2V充分利用了CNN和bi-LSTM模型,从而获得了最佳的性能。该方法利用CNN提取组合DNA序列的高阶抽象特征,并利用bi-LSTM捕获序列的双向语义依赖关系。
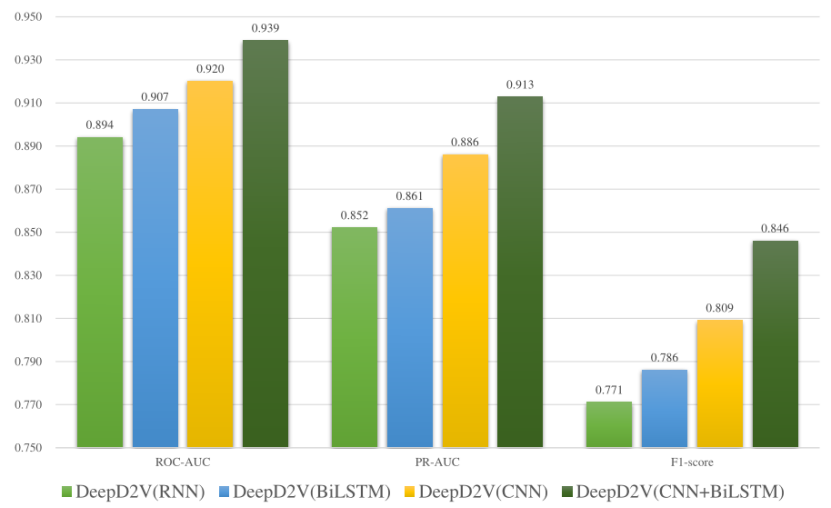
图7.不同网络结构的比较为了更好地评估DeepD2V的性能,本文在50个ChIPseq基准数据集上比较了DeepD2V与三种基于深度学习的模型,包括DeepBind、DanQ和WSCNNLSTM。所有的竞争方法都使用它们的作者发布的源代码运行。此外,文章通过交叉验证优化这些竞争方法的超参数,使模型在测试集上达到最佳性能。这使得与本文的方法的性能比较是客观的。图8显示了50个ChIP-seq数据集上的AUCs和f1得分。散点图中的每个点表示同一数据集上两种方法的性能度量。位于对角线左上角的点表明,DeepD2V模型的性能优于相应的方法。从图8a可以看出,DeepD2V在f1评分和AUC指标上的表现明显高于DeepBind。这种优势可能归因于组合输入,并增加了一个RNN层,以捕获图案的长期依赖性,这在DeepBind中没有应用。此外,根据f1评分和AUC指标,DeepD2V的表现明显优于DanQ,如图8b所示。该结果证明dna2vec的分布式密集表示优于单热编码稀疏表示。如图8c所示,基于f1评分和AUC指标,DeepD2V的性能优于WSCNNLSTM,说明联合输入比原始DNA序列更能提取有效的基序特征。总之,DeepD2V的性能优于其他三种方法,在50个体内ChIP-seq数据集上的性能指标f1评分、PR AUC和ROC AUC均达到了最先进的性能。这三个指标在50个数据集上的平均得分如图9所示。不同数据集上的ROC AUC值如表2所示。这些结果一致表明,DeepD2V的性能优于DeepBind、DanQ和WSCNNLSTM。
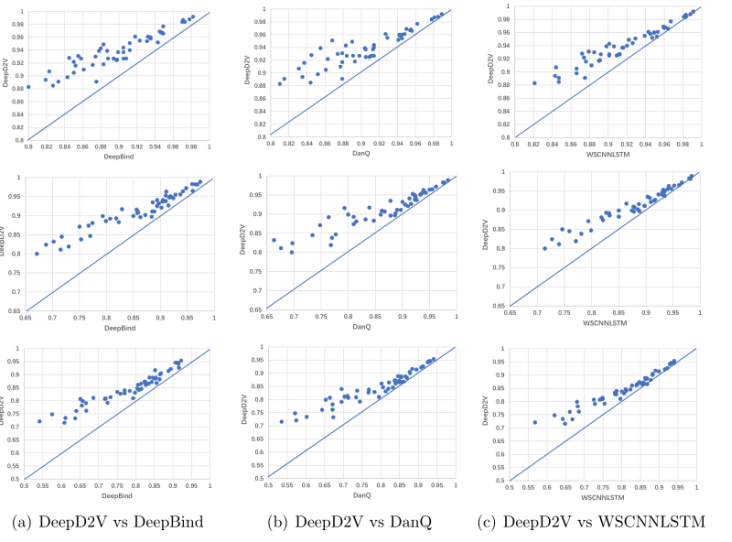
图8.DeepD2V与竞争方法的比较
6.** 总结**本文提出了一种基于深度学习的蛋白质-DNA结合预测新框架。首先,考虑反向互补序列对预测模型进行训练。本文假设蛋白质可能与逆序列或互补序列而不是原始序列结合。因此,本文考虑原始序列、反向序列、互补序列和反向互补序列的各种组合。本文的实验结果证明了组合序列有助于提取序列的特征。其次,在DeepD2V中采用词嵌入算法训练k-mers的分布式表示;分布式表示改进了后续的分类任务和特征学习。此外,提出了一种新的基于深度学习的框架,该框架以组合DNA序列为输入,同时使用CNN和bi-LSTM提取高阶和长时间特征。与其他单纯基于序列特征的深度学习方法相比,DeepD2V在蛋白质- dna结合预测方面具有更好的鲁棒性和性能。