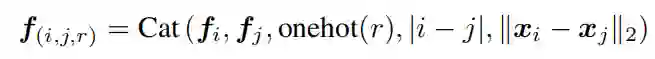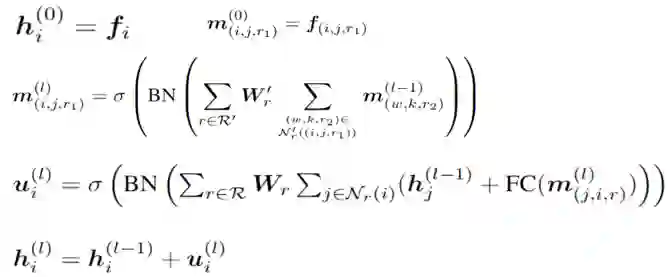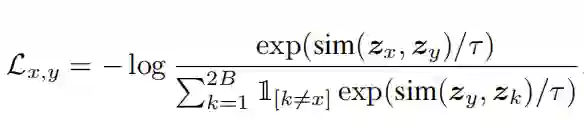作者 | 李政毅
审核 | 黄 锋

今天给大家介绍的是2022年Mila、蒙特利尔、剑桥大学等机构发表的一篇论文:"Protein Representation Learning by Geometric Structure Pretraining"。作者开发了一个通用的蛋白质编码器(几何感知关系图神经网络(GearNet)),它通过添加不同类型的结构边来编码空间信息,然后在蛋白质图上执行关系消息传递(还加入了一种边缘消息传递机制来增强蛋白质结构编码器。作者提到,这是首次尝试将边缘级别的消息传递结合到蛋白质结构理解中,也是蛋白质结构建模领域的新颖之处,以前的工作只考虑残基或原子之间的消息传递)。利用多视图对比学习和不同的自预测任务来预训练蛋白质图编码器。最终实验结果表明,模型在不到100万个样本上的训练获得了与在100万或10亿级数据集上预训练的最先进的基于序列的编码器相媲美甚至更好的结果。
1 研究背景
以前的工作试图学习基于不同形式的蛋白质的蛋白质表示,包括氨基酸序列和蛋白质结构。这些工作的共同目标是学习信息丰富的蛋白质表征,这有助于各种下游应用,如预测蛋白质功能和蛋白质-蛋白质相互作用。 尽管基于序列的方法很有效,但是蛋白质结构才是已知的蛋白质功能的直接决定因素。为了更好地利用这一关键的结构信息,已经提出了一些基于结构的蛋白质编码器。然而由于蛋白质结构的稀缺性,这些编码器往往是为专门的任务而设计的,因此不清楚在多大程度上适用于其他任务。并且上述蛋白质结构稀缺的原因,直到最近,开发利用3D结构的通用蛋白质编码器的尝试还很少。 基于深度学习的蛋白质结构预测方法已经有了最新进展(AlphaFold),现在可以有效地预测大量蛋白质序列的结构。在这一发展的推动下,作者开发了一个通用的蛋白质编码器(几何感知关系图神经网络(GearNet))。
2 模型构建
首先明确模型的目标:给定蛋白质结构,模型旨在学习编码其空间和化学信息的表征。
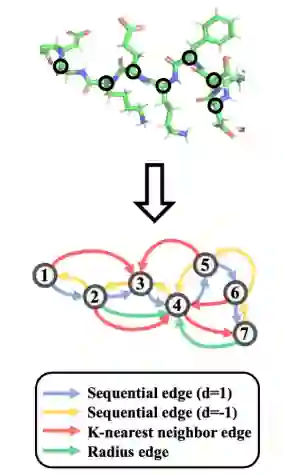
2.1 Protein graph的构建
,其中表示边的类型,每个节点代表一个残基的 α碳,所有节点的 坐标为 。
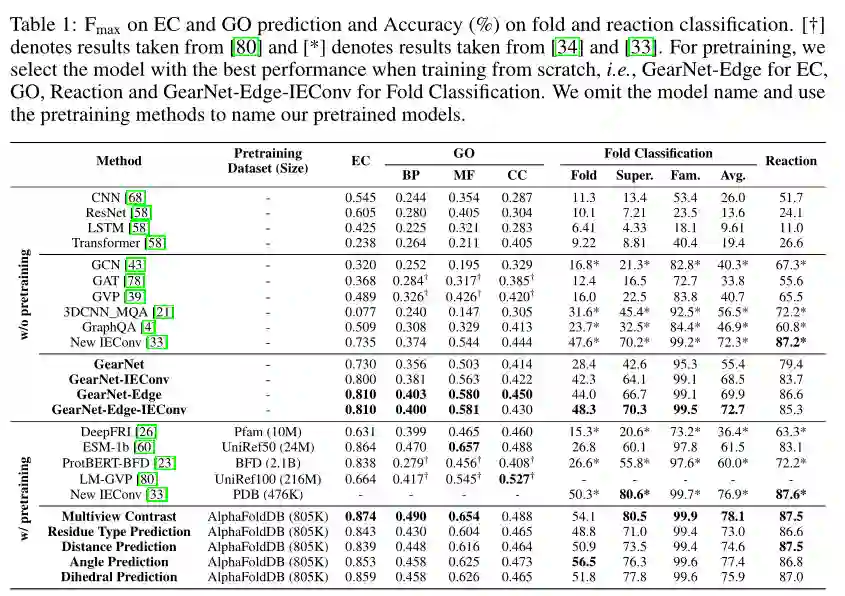
「将三种不同类型的空间边添加到蛋白质图中」
(这些边类型可以反映不同的几何特性,它们共同产生了蛋白质的全面特征):
- :如果第i个残数和第j个残数之间的序列距离低于预定阈值,即,则添加边。
- :当i和j之间的欧式距离小于一个阈值时,添加边。
- :由于不同蛋白质的空间坐标尺度可能不同,一个节点也将基于欧式距离与其k个最近的邻居相连。这样,保证了不同蛋白质图之间的空间边密度具有可比性。 由于作者对序列中相互靠近的残基之间的空间边不感兴趣,所以进一步对后两种边过滤。具体地说,对于连接第i个和第j个残基的半径或边,如果它们之间的序列距离小于设定阈值,即,则将后两种边删除。
进一步说明:
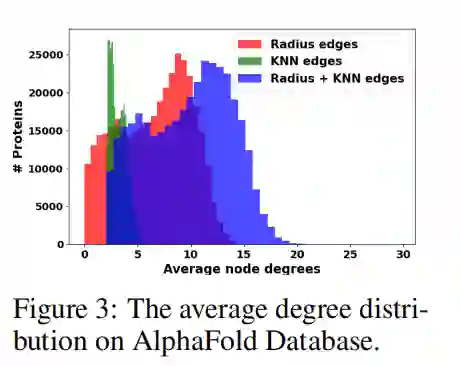
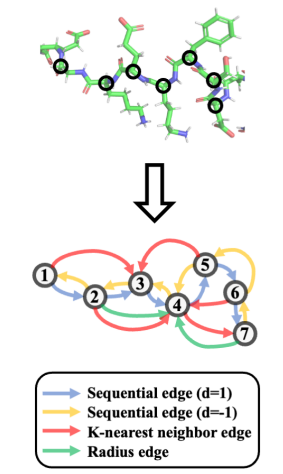
在这里,作者通过统计来解释半径和 KNN 边的必要性。这两种边导致非常不同的度分布。绘制了 数据库中蛋白的平均度分布,如下图:
如上图可以说明,若只考虑 KNN 边,蛋白质图中的节点度数接近于一个常数,这使得难以捕捉那些残基之间相互作用密集的区域; 如果只考虑半径边,那么将有大约 15,000 个平均度数低于 2 的蛋白质。在这些稀疏图中,预训练不能有效地捕捉结构信息。这种稀疏性很难通过调整半径截止来克服,因为不同蛋白质的平均距离有不同的尺度。通过简单地结合两种边,可以克服这些问题。
2.2 Node and edge features
节点:
大多数以前为设计的基于结构的编码器使用许多化学和空间特征,其中一些难以获得或计算耗时。相比之下,作者只使用one-hot 作为节点特征,表示为
边:
边的特征是两个端节点的节点特征;边类型的one-hot编码;它们之间的序列和空间距离的一个串联:
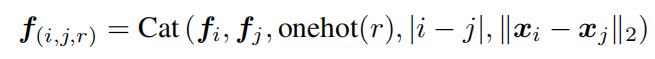
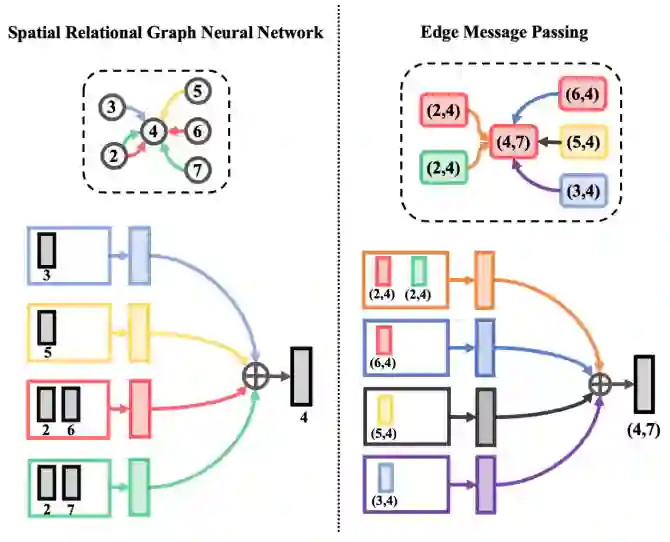
然后,应用关系图卷积层。类似的消息传递层可以应用于边图以提高模型的质量。分别显示了节点和边(,)的更新迭代,如下图:
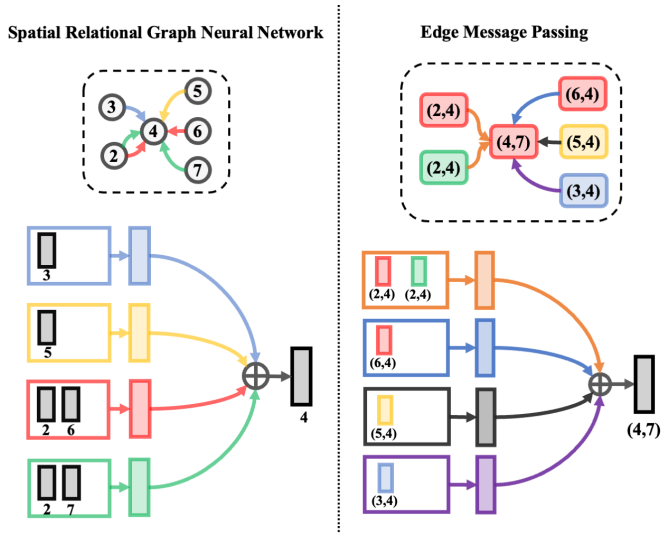
说明:
图中,Edge Message Passing需要首先构造一个边之间的关系图,,。图中的每个节点对应于原图中的一条边,将原图中的边,,链接到边,,当且仅当且。这条边的类型由,,和,,之间的角度确定。将,π离散为8个区间,并使索引作为边缘类型。 消息传递的具体公式如下:
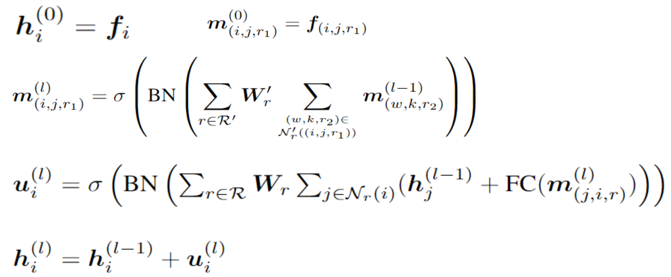
3 预训练模型
作者遵循了两种流行的自监督学习框架:对比学习和自预测
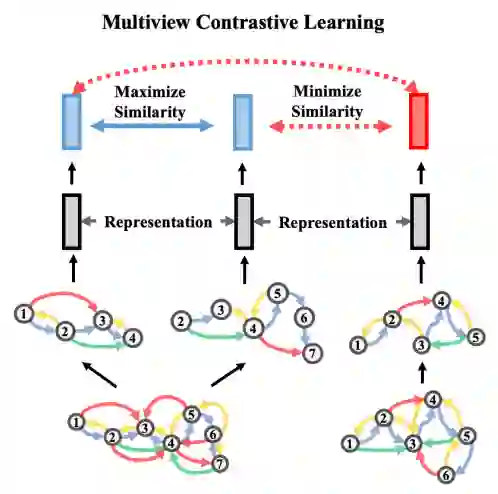
对比学习:
考虑两个不同的剪裁函数生成蛋白质的不同视图,随机选择以下两种等概率变换中的一种来应用于裁剪蛋白质图,如下图所示: 子序列:随机抽样一段连续的蛋白质序列,并从蛋白质图中取出相应的子图。 子空间:随机抽取一个残基作为中心,选择指定半径球内的所有残基。
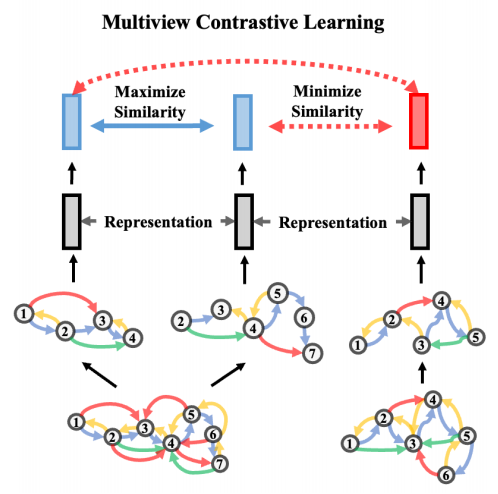
然后,分别计算视图的表示和,应用两层将表示映射到和。对于正对和,将来自同一批次中的其他蛋白质的视图视为负对(目标是最大化来自同一蛋白质的视图,同时最小化来自不同蛋白质的视图之间的相似性),定义:
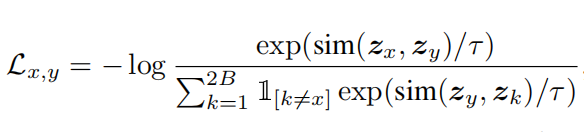
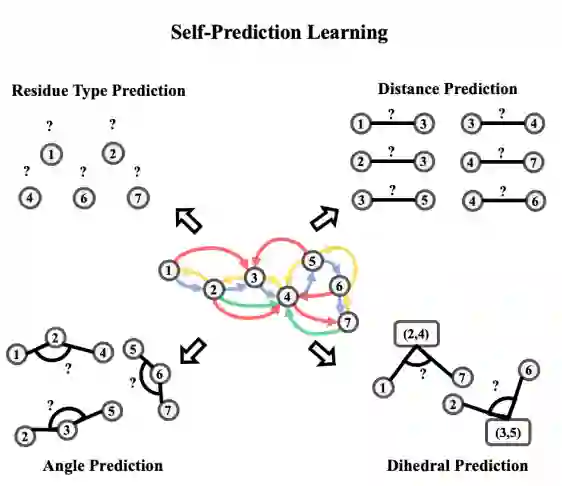
自预测:
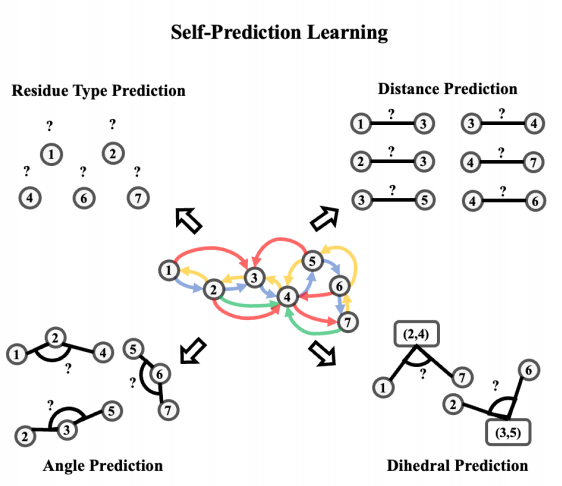
包括四个任务,残基类型预测,距离预测,角度预测,二面角预测,具体如下图所示:
4 实验
在四个标准的下游任务上对模型进行评估,包括:Enzyme Commission (EC) number prediction,Gene Ontology (GO) term prediction,Fold classification,Reaction classification。结果如所示:
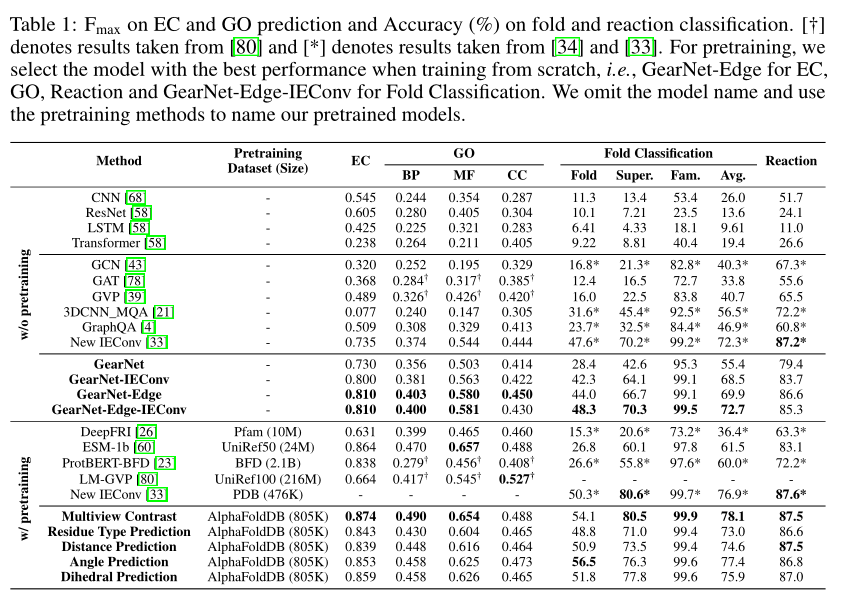
可以发现:
1)作者的基于结构的编码器在8个数据集中的7个上无需预训练即可超越所有基线模型; 2)预训练的基于结构的编码器的性能与预先训练了更多数据的基于序列的编码器相当,甚至更好,最后两块显示了基于序列的预训练模型和基于结构的模型之间的比较。作者的模型是在具有少于一百万个结构的数据集上进行预训练的,而所有基于序列的预训练基线模型都是在百万或十亿级的序列数据库上进行的预训练。虽然数据量少了一个数量级,但获得类似的甚至更好的结果。 3)此外,考虑到基于序列的模型在折叠分类上的表现不佳,作者的模型是唯一能够在所有四个任务上都获得良好性能的模型。这再次表明了基于结构的预训练模型在学习蛋白质表示方面的潜力。