PNAS | 深度学习指导优化具有广泛中和作用的针对 SARS-CoV-2 变体的抗体
文章简介
本文于2022年3月1日发表在PNAS,清华大学医学院张林琦教授团队、UIUC彭健教授团队等开发了一种深度学习方法来重新设计互补决定区 (CDR) 以靶向多种病毒变体,并获得了一种广谱中和 SARS-CoV-2 变体的抗体。


要解决的问题:
目前,新冠病毒正在全球广泛传播,导致数亿人确诊感染和数百万人死亡。病毒变异和逃避人类免疫系统和中和抗体的能力仍然是抗病毒药物和疫苗开发的障碍。许多获批使用的中和抗体对SARS-CoV-2 变体的活性降低或完全丧失。
目前传统的抗药药物获得一般采用基于实验的筛选方法配合体外亲和力成熟的方法,例如使用展示技术与随机诱变技术,已被证明可以改善抗体与靶蛋白的结合,但这种方法既费时又费力。针对一种特定变体的目标优化也可能导致对其他变体的中和活性丧失。因此,迫切需要针对多种变体的广谱中和抗体。
病毒还在不断变异,例如B.1.617 谱系,也称为 Delta 变体,在 RBD 中包含两个促进病毒逃逸的突变(L452R 和 T478K),L452R 突变的较长侧链可能对抗体识别产生了空间干扰。Omicron甚至出现更多的突变位点来逃避免疫识别,导致中和抗体和疫苗的失效。毫无疑问,迫切需要开发和优化新的中和抗体,甚至提前设计可以中和更多高风险突变病毒的广谱抗体药物。
结果:
作者团队开发了一种几何深度学习算法,可有效增强抗体亲和力,实现了针对不同新冠变体的更广泛和更有效的中和活性。作者团队以抗体 P36-5D2出发,通过计算+实验多轮优化该抗体的互补决定区 (CDR) 序列,将其中和Delta的效力提高 10 到 600 倍。
作者团队开发了一种基于注意力的几何神经网络架构,从三维蛋白质复合结构中学习突变对蛋白质-蛋白质相互作用的影响(如图)。具体来说,对于蛋白质复合物中的每个残基,网络首先通过注意力机制识别其他残基的重要性,并从中提取信息,包括空间接近度和物理化学性质。为了评估突变的影响,首先通过在突变位点周围重新包装侧链来预测突变蛋白复合物的结构,并使用网络编码野生型 (WT) 和突变复合物以获得 WT 和突变体嵌入。然后,额外的神经网络层比较两个嵌入,以预测突变的影响。
模型:
考虑到效率问题,对复合物建模时只考虑界面上残基,故使用最接近另一条链的48个残基来训练模型。残基特征和残基对的信息通过几何注意力网络编码,每个残基i的坐标被输入网络,pi为原子坐标,ti为Cα坐标,Ri为朝向并通过MLP编码为hi。残基i和残基j之间的残基对信息为向量zij,加上距离信息wi三部分共同连接成mi并由多头注意力网络迭代更新。最终每个残基的特征向量编码了它与周围残基的相互作用,可以结合Rosetta计算的复合物能量项特征,通过反对称网络计算野生型和突变体的差异,评估结合亲和力的变化。
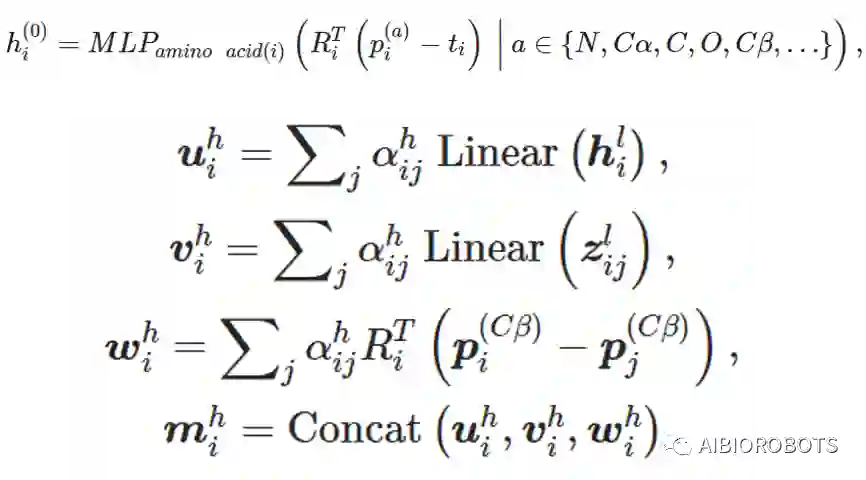
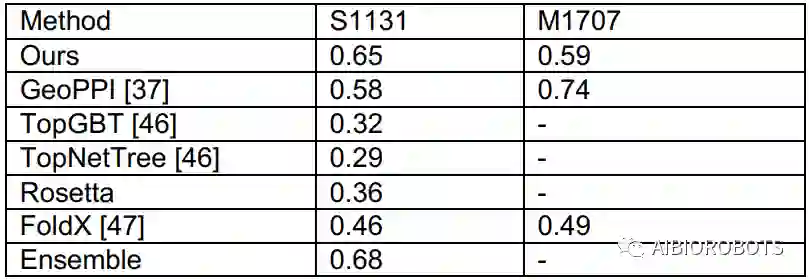
本模型使用目前最大的抗体-抗原结合亲和力数据集SKEMPI V2.0,共包含342个复合物和5318个突变,该数据集被平均分成不互相重复的五个子集在几何网络上进行五折交叉验证。在数据集S1131(单点突变)和M1707(多点突变)上与其他模型预测的ΔG值与实际ΔG之间的皮尔逊相关系数如上图所示,可以认为本文模型预测结果与实验有较好相关性。其中GeoPPI为SOTA的基于图神经网络和梯度提升树的结合亲和力模型,集成模型为本文模型、GeoPPI和Rosetta Cartesian ΔG的平均结果,并被用作最终筛选突变的标准。
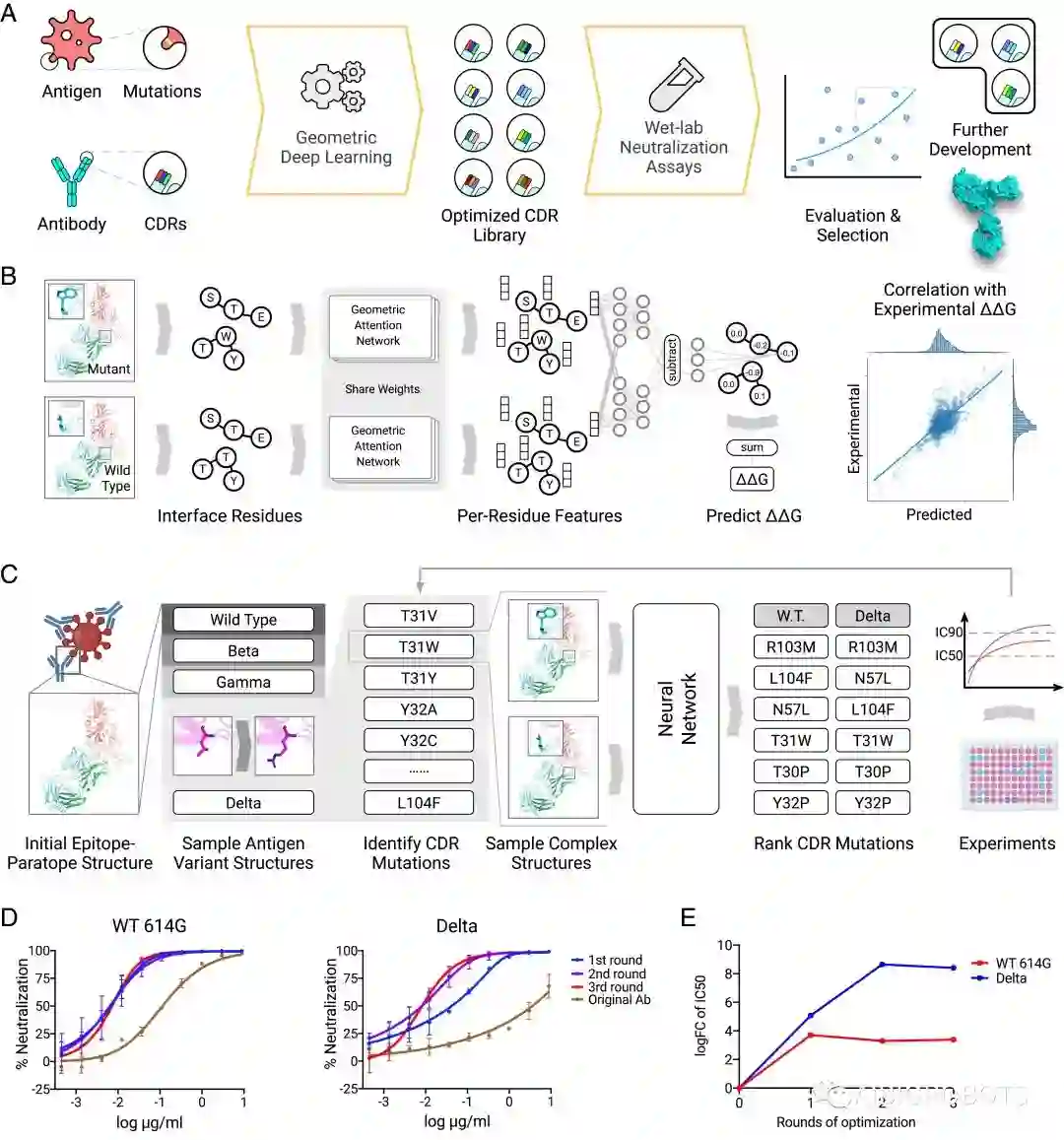
A:抗体优化的计算+实验反馈循环。
B:WT 复合体和突变的复杂结构使用共享的几何注意网络进行编码,然后通过比较两个复合物特征的网络预测 ΔΔG 来评价突变效果。
C:首先基于结构进行单位点突变,然后预测ΔΔG改善效果,最后实验检测将具有中和效果的突变进行组合。
D:优化提高了对 SARS-CoV-2 和 Delta 变体的中和能力。
E:IC 50相对于原始抗体的对数倍数变化。
为了提高 P36-5D2 对 Delta 变体的中和活性,基于结构与对其他毒株的活性评价生成了一个抗体 CDR 的计算机突变库,通过训练有素的几何神经网络进行排序,这样它们不仅应该提高抗体与 Delta RBD 的结合,而且还应该保持与其他变体 RBD 的结合。通过假病毒中和试验对 P36-5D2 进行了总共四轮优化,以选择具有最佳中和效力和广度的优化抗体。
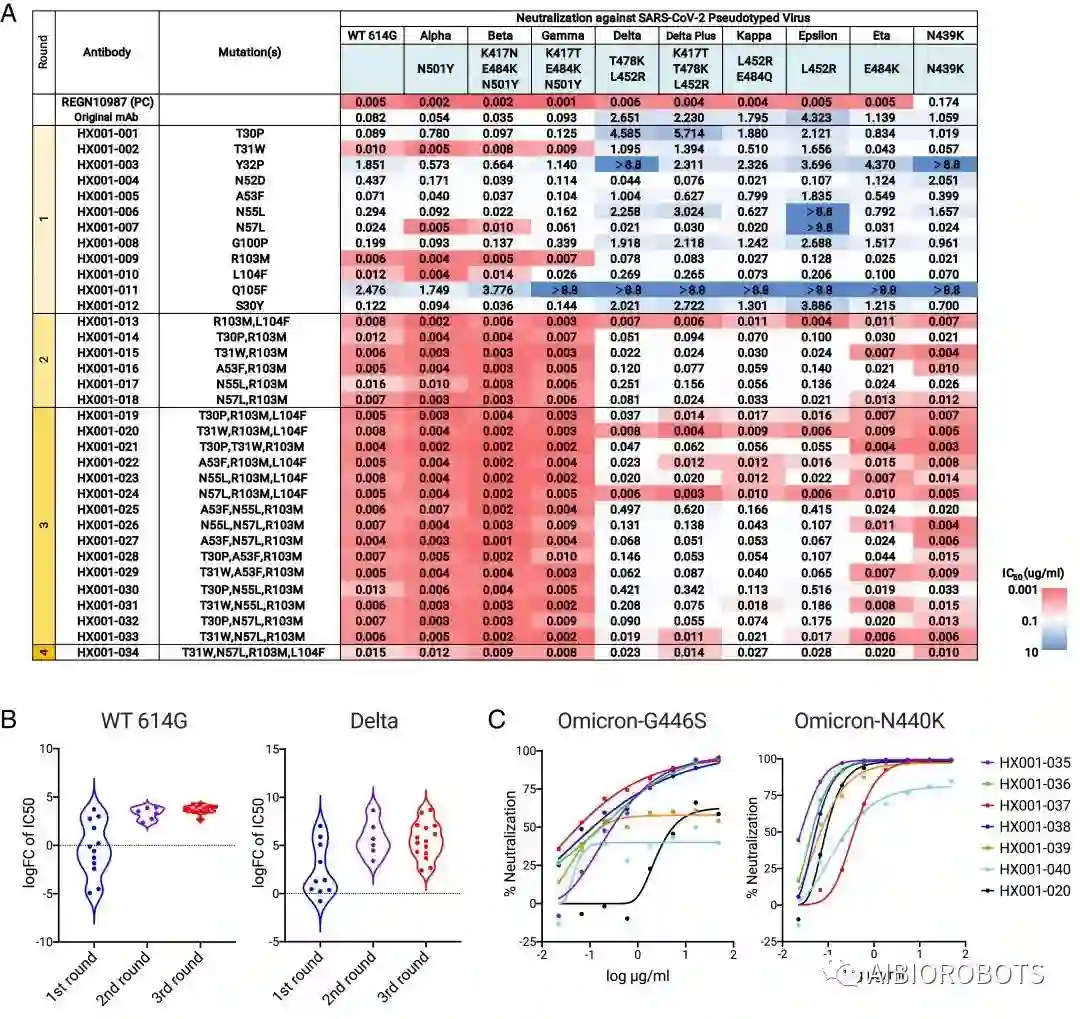
第一轮优化结果
突变HCDR3中的R103M显著提高了P36-5D2对所有10种假型病毒的中和活性,平均半数最大抑制浓度(IC 50 )达到0.038 μg/mL。HCDR1 中的T31W、HCDR2 中的 A53F 和 N57L 以及 HCDR3 中的 L104F 四种突变也提高了整体中和活性,平均 IC为 50分别为 0.479 μg/mL、0.547 μg/mL、0.025 μg/mL 和 0.104 μg/mL。
第二轮优化结果
HX001-013(R103M 和 L104F)和 HX001-015(R103M 和 T31W),其平均IC 50值分别增加到0.007μg/mL和0.013μg/mL(上图)。HX001-013 有一个 IC 50类似于参考抗体 REGN10987,在中和 N439K 变体方面比 REGN10987 更有效。
第三轮优化结果
HX001-020(R103M、T31W 和 L104F)、HX001-024(R103M、N57L 和 L104F)和 HX001-033(R103M、 T31W 和 N57L) 的平均 IC 50分别为 0.006 μg/mL、0.006μg/mL 和 0.010μg/mL(上图)。HX001-020和HX001-024展示IC 50值通常与参考抗体 REGN10987 相似,并且在中和 N439K 突变体方面优于 REGN10987。
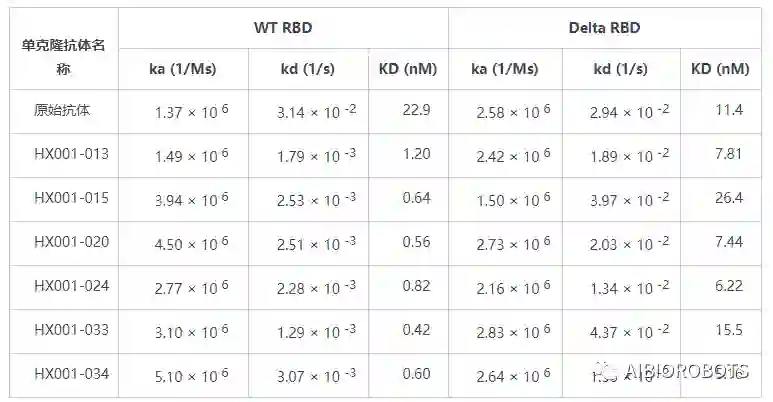
对于SARS-CoV-2 WT RBD,优化后的抗体的解离常数 (K D ) 为 1.2 nM 至 0.42 nM,比原始 P36-5D2 抗体强 20 至 50 倍。虽然针对 WT RBD 的原始抗体的解离率 (k d ) 值约为 10 -2,但六种优化抗体的解离率 (k d ) 值均达到了 10 -3,意味着更长的半衰期结合期和更高的结合稳定性。值得注意的是,优化抗体与 SARS-CoV-2 Delta RBD 的解离常数为 6.22 nM 至 26.4 nM,是 P36-5D2 的两倍或相似。
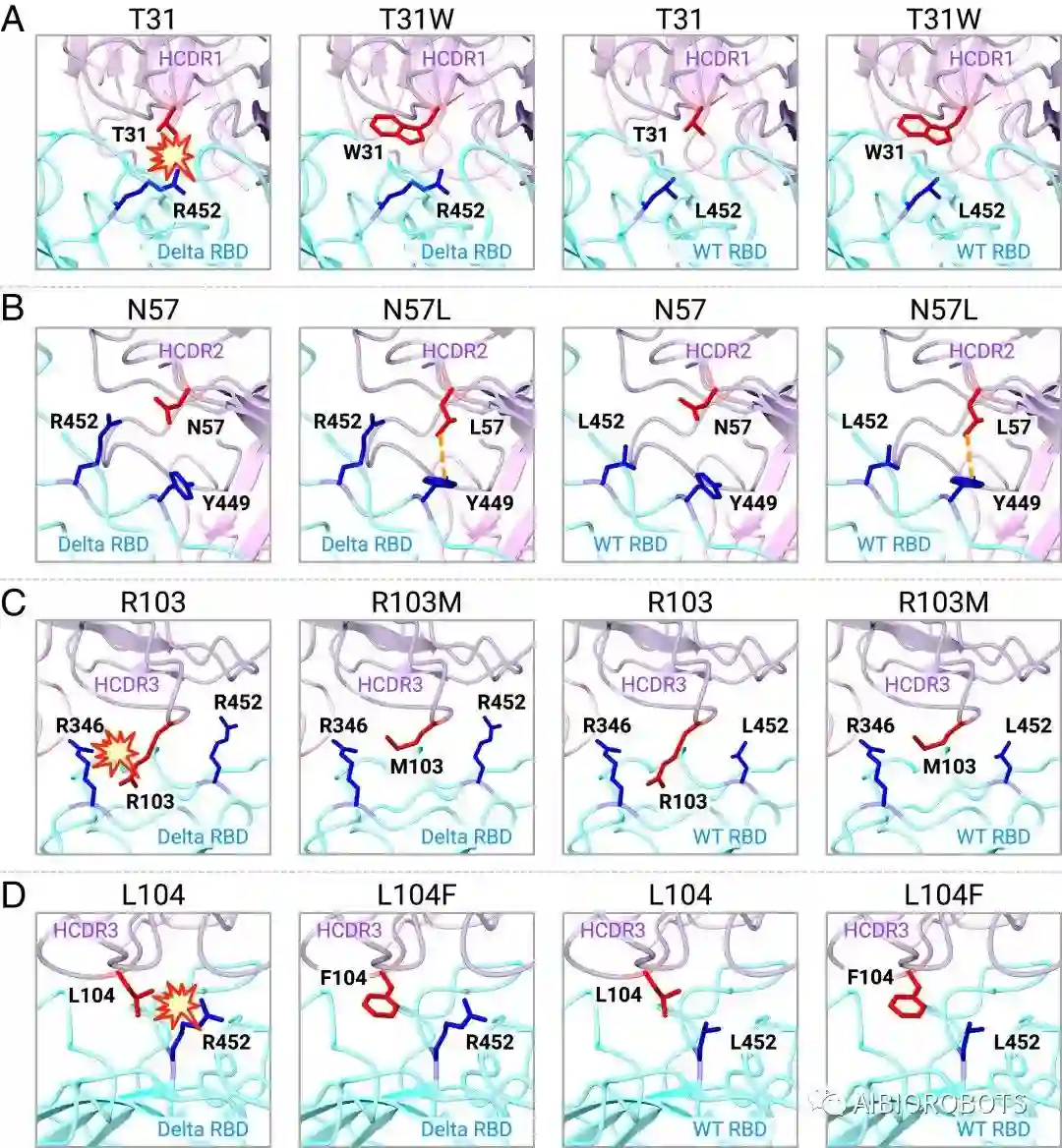
使用 Rosetta 来预测 WT/Delta RBD 的结构,其中抗体携带四个关键的单突变:T31W、N57L、R103M 和 L104F。如图所示,原始抗体 HCDR1 上的T31 可能与 Delta RBD 上的 R452 侧链发生空间冲突,因为两个侧链上的重原子之间的最小距离在 4 Å 以内。W31 代替 T31 后,空间位阻消失。HCDR2上用 L57 取代 N57 后,观察到 L57 的侧链和 Y449 之间的新相互作用,可能解释了这种突变增强亲和力的原因。HCDR3 上的 R103 在空间上非常接近 Delta RBD,这也可能导致与 R346 的空间冲突。由于 R103 和 R346 都具有很长的侧链并带有正电荷,因此两者之间的接近可能会引入强烈的排斥力,可能会大大降低抗体与抗原之间的结合。替换具有更小的侧链的 M103 可能解释了对 Delta 变体的极大改进的中和作用。HCDR3 上用 L104 代替 F104 也可能消除与 Delta RBD 上的 R452 的潜在空间冲突,从而改善结合。
2021 年 11 月,南非出现了一种新的 SARS-CoV-2 变体 Omicron。Omicron 总共携带 36 个刺突蛋白突变和 15 个 RBD 突变,极大地改变了 RBD 的免疫原性,并导致许多中和抗体失效。通过结构分析,作者团队发现G446S突变位于抗体P36-5D2的表位上,而N440K在表位附近(5埃以内)。因此通过深度学习方法来进一步优化抗体序列 HX001-020。鉴定的最佳突变包括LCDR3中的N92F (HX001-035)、G93M (HX001-036)、Y94G (HX001-037) 和Y74D (HX001-038)。一个双突变体HX001-039 (N92F/Y94G) 和一个三重突变体 (N92F/G93M/Y94G)。优化的抗体在中和 N440K突变株方面比 HX-020 高 1 到 3 倍,在 G446S 突变株上强 20 到 100 倍。
评论
从文章中的案例来看这个方法的优势在于可以在非常短的时间内对抗体有针对性的进行亲和力成熟,相对于传统的实验方法可以获得广谱的中和抗体。但是需要复合物结构作为基础,可能应用会受到一些限制。在新冠这个应用案例上来看如果可以结合新流行毒株突变位点的预测模型来提前设计中和抗体可能会很有趣。对于抗体设计与优化这个领域我们很高兴看到AI已经可以发挥一定作用,也希望看到更多的应用案例。目前多家公司已经宣布在AI抗体/抗原设计领域布局,有理由相信不久的将来会有更多的新技术与新成果会诞生。
参考文献:
https://doi.org/10.1073/pnas.2122954119
Geoppi:
X. Liu, Y. Luo, P. Li, S. Song, J. Peng, Deepgeometric representations for modeling effects of mutations on protein-proteinbinding affinity. PLoS Comput. Biol. 17, e1009284 (2021).
代码:
https://github.com/HeliXonProtein/binding-ddg-predictor



