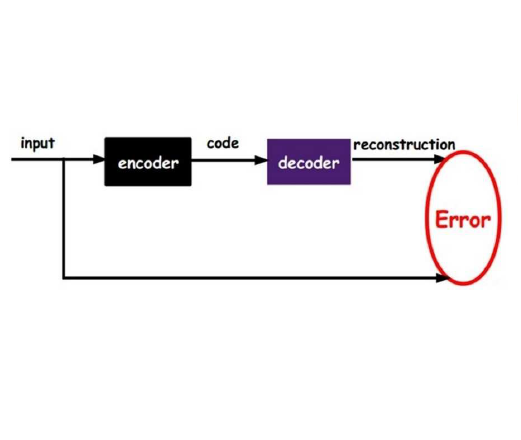
Single-cell RNA sequencing (scRNA-seq) has revolutionized our ability to study individual cellular distinctions and uncover unique cell characteristics. However, a significant technical challenge in scRNA-seq analysis is the occurrence of "dropout" events, where certain gene expressions cannot be detected. This issue is particularly pronounced in genes with low or sparse expression levels, impacting the precision and interpretability of the obtained data. To address this challenge, various imputation methods have been implemented to predict such missing values, aiming to enhance the analysis's accuracy and usefulness. A prevailing hypothesis posits that scRNA-seq data conforms to a zero-inflated negative binomial (ZINB) distribution. Consequently, methods have been developed to model the data according to this distribution. Recent trends in scRNA-seq analysis have seen the emergence of deep learning approaches. Some techniques, such as the variational autoencoder, incorporate the ZINB distribution as a model loss function. Graph-based methods like Graph Convolutional Networks (GCN) and Graph Attention Networks (GAT) have also gained attention as deep learning methodologies for scRNA-seq analysis. This study introduces scVGAE, an innovative approach integrating GCN into a variational autoencoder framework while utilizing a ZINB loss function. This integration presents a promising avenue for effectively addressing dropout events in scRNA-seq data, thereby enhancing the accuracy and reliability of downstream analyses. scVGAE outperforms other methods in cell clustering, with the best performance in 11 out of 14 datasets. Ablation study shows all components of scVGAE are necessary. scVGAE is implemented in Python and downloadable at https://github.com/inoue0426/scVGAE.
翻译:暂无翻译