2022年9月,安进药物产品技术部的Prashant Agarwal等人在Drug Discov Today发表文章Trends in small molecule drug properties: A developability molecule assessment perspective,分析了1900-2020年期间,FDA批准的口服小分子药物的特性变化。本文可为药物研发团队的开发决策提供参考。

概要
分子的可开发性评估是整个生物制药行业的关键能力。 然而,大多数描述理化性质趋势的文献资料都是从药物化学的角度来写的,目的是为了确定具有更好的ADMET特征的分子,其中未能描述这些性质对CMC(chemistry manufacturing controls)的影响。为了系统地考察理化性质对CMC发展的相应影响,本文对药物再利用中心数据集中的分子进行了全面分析。 分析结果显示,1900-2020年期间,FDA批准的口服分子中,H-键供体(H-bond donor,HBD)数量随着时间变化保持不变(似乎是唯一保留不变的性质)。Mw/PSA/HBA持续呈上升趋势,平均cLogP只是在过去十年中有所增长。按靶点类别排列的数据集表明,可以根据分子的靶点类别来预测或实施某些制剂和给药策略。 简介
分子可开发性评估(Developability molecule assessment, DevMA)或早期候选评估(early candidate assessment, ECA)是药物发现项目的关键部分。药物分子不尽如人意的理化性质,如高亲油性,会直接影响药物的性质(如溶解性、毒性、疗效和可制造性),导致临床上的损耗率升高。 本文分析了来自药物再利用中心的分子的物理化学性质(这个公开的库是一个小分子化合物的综合集合),并对其开发阶段、靶点蛋白、疾病领域和作用机制(MoA)进行了注释。该库于2017年首次推出,2020年4月进行了更新,包含7000个独特的分子。 本文从DevMA的角度对药物再利用中心数据集中的分子进行了深入分析,主要分析内容如下:基于药物理化性质与批准年份的数据,评估FDA批准的口服药物的时间-性质关系;基于理化性质与开发阶段的数据,评估影响整个制药业损耗的分子的理化性质;基于理化性质与药物靶点类别和/或亚类的数据,探讨药物的不同理化性质与其蛋白质靶点的性质和类型的关联(类似于基于靶点类别的药物发现)。 数据整理、分配和量化
药物再利用中心的分子的开发阶段数据被注释为临床前、I期、II期、III期、撤消或上市。 总共有7196个分子被纳入损耗分析,其中包括2341个临床前分子,2892个已上市分子,609个正在临床中的分子,1192个已中止的临床分子和162个临床状态无法核实的分子。 根据IUPHAR/BPS的药理学指南,靶点类别根据主要的治疗性蛋白质类别来分配。蛋白质被分为八类:激酶、其他酶(非激酶)、核激素受体(NHRs)、其他转录因子、G-蛋白偶联受体(GPCRs)、离子通道、其他运输工具和非传统靶点。亲油性的定义是将极性表面积(PSA)中固有的极性和H键项与分配系数(cLogP)相结合。亲油性的这一指标已被证明可以识别能够成功通过生物膜的分子。 理化性质与批准年份的关系
不随时间变化的分子特性是最重要的。对获得成功的概率至关重要的分子特性必须是恒定的。 本文考察了多个分子理化特性与批准年份的关系。 对1900年至2020年期间,FDA批准上市的口服分子进行的时间依赖性分析表明,根据10年的移动平均值,Mw随着时间的推移而增加,最大的跳跃出现在过去十年的2011-2020年。 不过,任何关于Mw及其对口服生物利用度的影响的讨论都需要结合最近FDA批准的口服赛马格列汀,这表明人体的口服吸收不能被限制在分子大小的单一参数上。应用处方方法,如使用口服渗透增强剂,可以帮助分子渗透到消化道内壁。
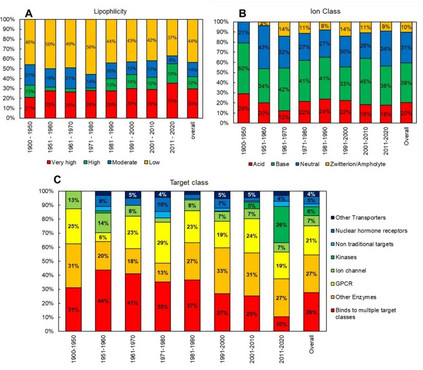
图1. 描述FDA批准的口服分子与理化性质的时间分析的折线图
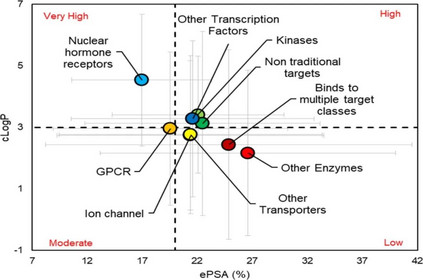
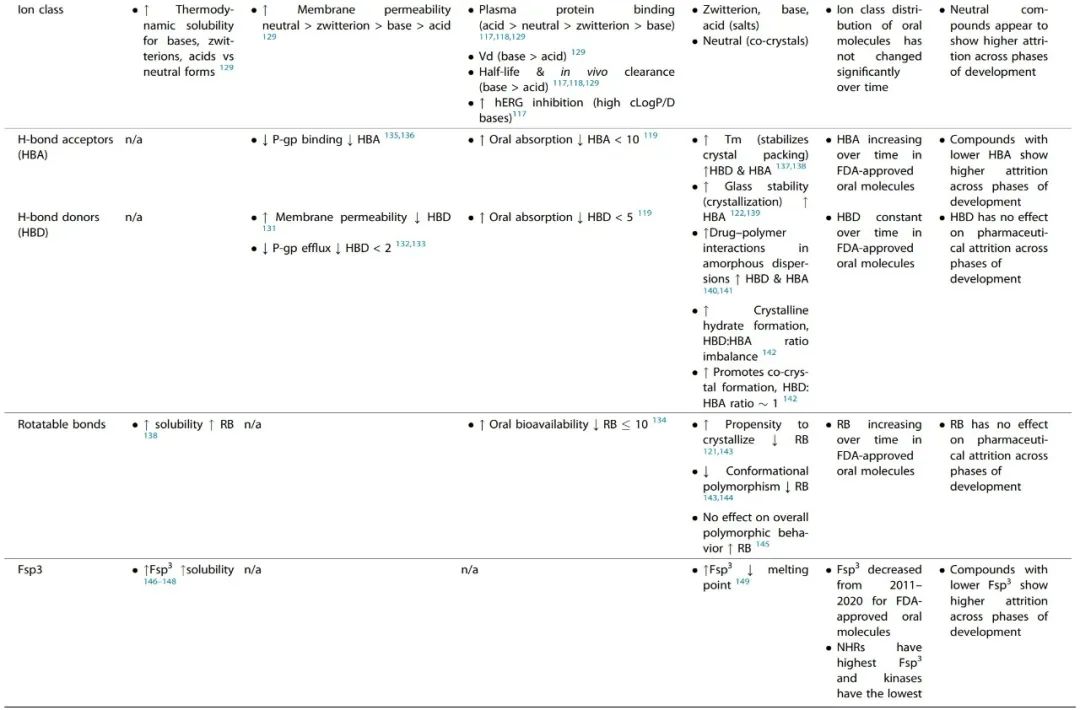
cLogP和cLogDpH6.5是传统的亲油性指标,这些特性在2010年之前保持相对稳定;但是,在过去十年(2011-2020 年),这些特性的平均值有了统计学上的显著增加(增加了 1 个对数单位)。根据cLogP和ePSA的定义,分子的总体亲油性在四象限图上呈现出轻微的上升趋势(图2a)。亲油性‘very high’和‘high’的分子比例从2001-2010年的41%增加到2011-2020年的54%。 然而,需要认识到四象限图在亲油性分类方面的局限性;因为每个象限涵盖了一个广泛的性质空间,而简单地将一个类别分配给一个分子子集是无法捕捉到特定象限内的变化的。
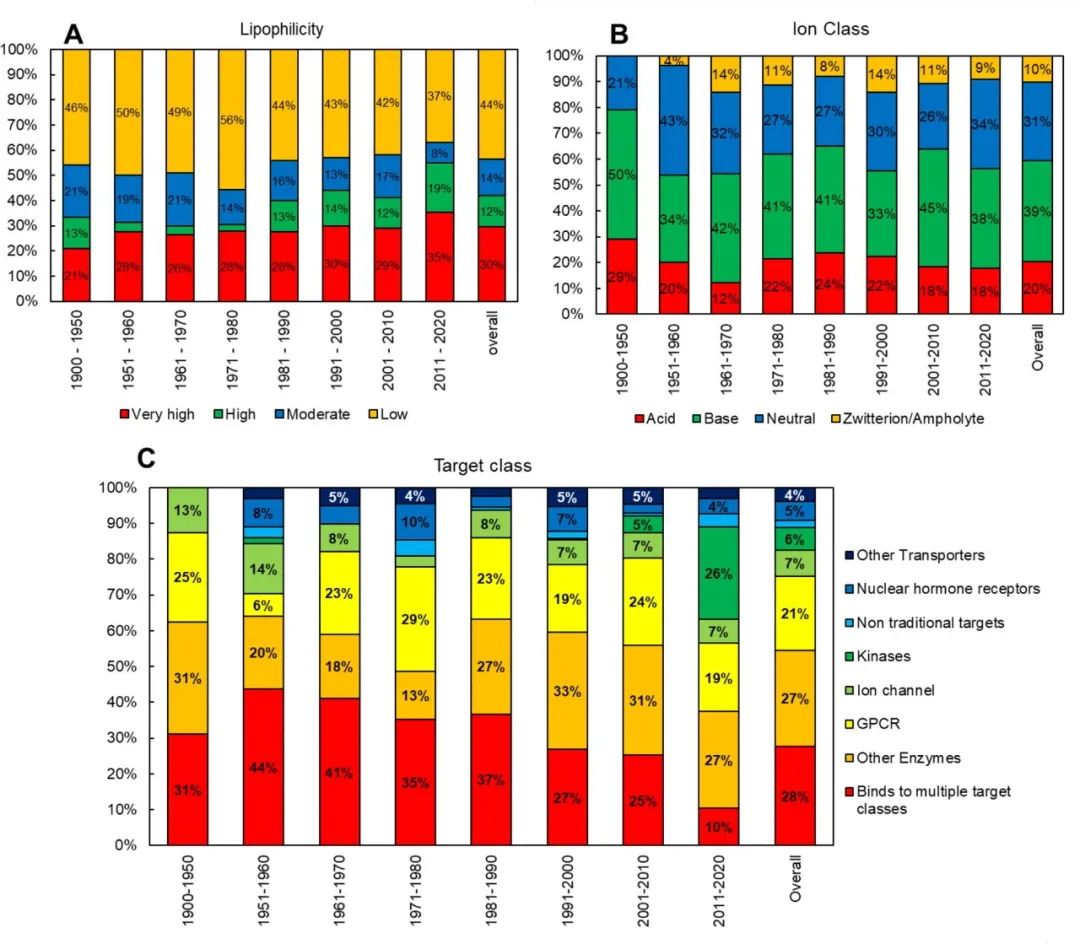
图2. 分子亲油性(a)、离子类别(b)和靶点类别(c)的柱状图,作为FDA批准的口服分子的函数(带批准年份) 唯一经得起时间考验的参数似乎是HBD(H-bond donor)。随着时间的推移,HBD的平均数量没有出现统计学上的明显变化。 Fsp3有时被用作分子复杂性和药物可能性的描述符,从2001年至2010年的平均0.45下降到2011年至2020年的0.37,具有统计学意义。这种下降的一个可能的原因是过去十年中批准的结合激酶的分子比例很高(图2c)。 图3显示了FDA批准的口服分子的Mw与cLogP的关系图,该图为10年的移动平均值。在过去的十年中,可以看到Mw和clogP的明显跳跃,平均Mw增加了∼100 Da,cLogP增加了∼1.5对数单位。2011年至2020年期间Mw和cLogP的变化可能表明,由于遗传学、组合化学和HTS方面的关注和进步,新发现的分子的理化性质发生了系统性变化。
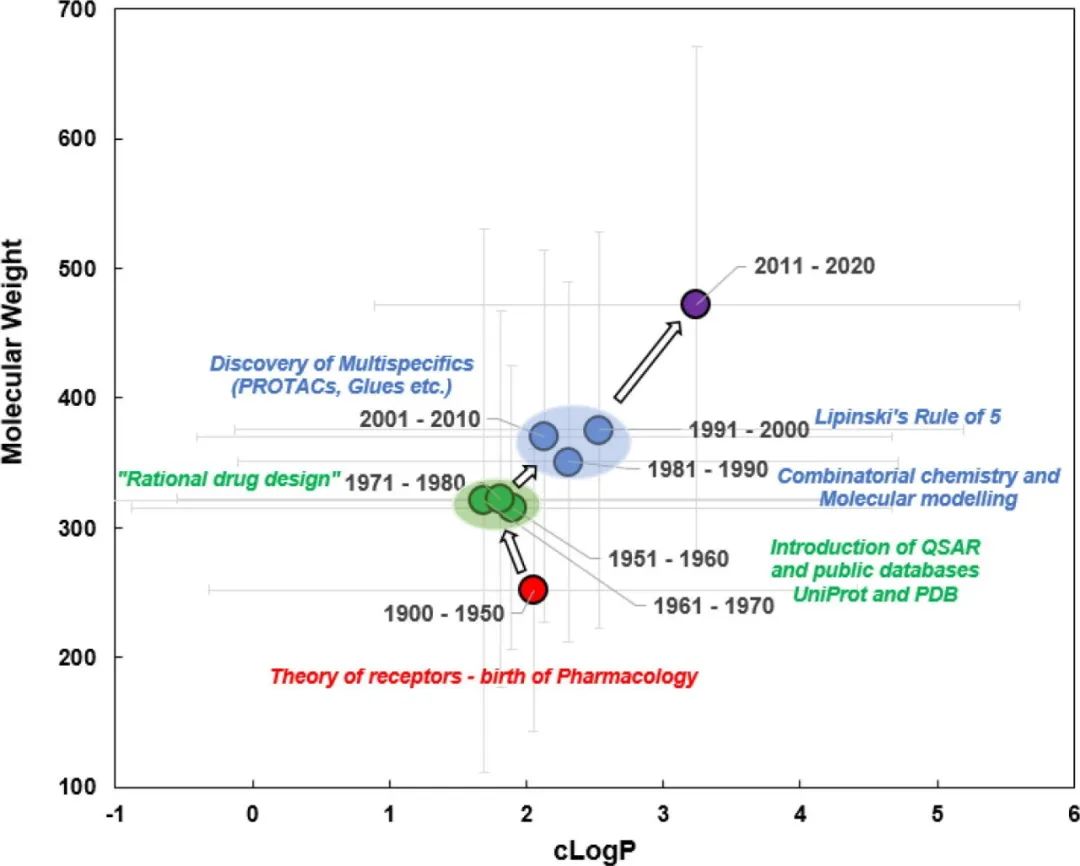
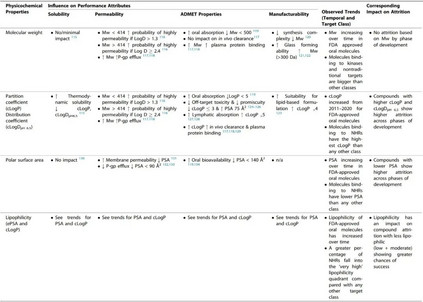
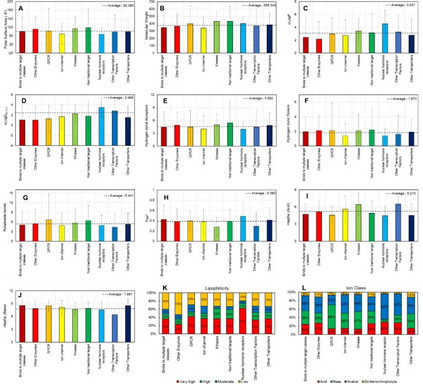
图3. 美国FDA批准的口服分子的cLogP与Mw的散点图(10年动态平均) 为了便于讨论,本文将从理化性质和性能性质的角度对观察到的趋势进行审查。性能性质被定义为受计算的物理化学描述符(如溶解性、渗透性、ADMET特性、可制造性)变化影响的特性。 表1总结了观察到的理化描述符随时间、损耗和靶点类别变化的趋势,并评估了它们对关键性能性质的影响。 表1. 理化性质及其与关键性能参数(溶解性、渗透性、ADMET特性和可制造性)的关系
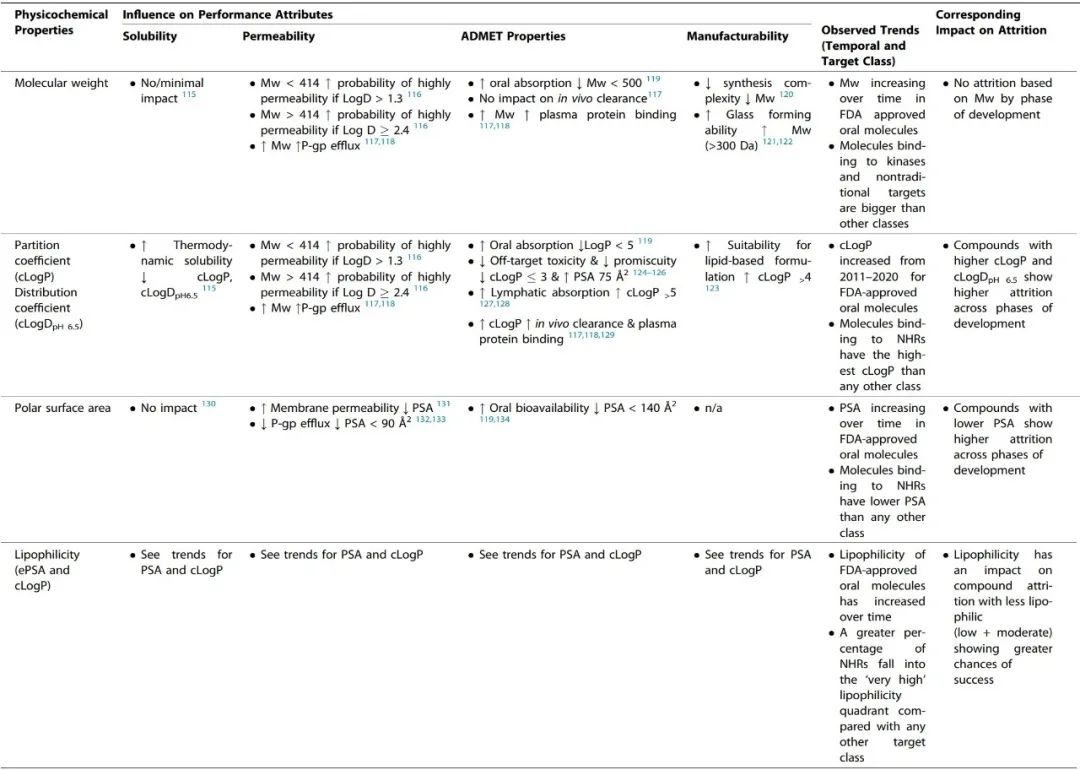
溶解度
Yalkowsky开发的一般溶解度方程提供了一个工具,用于根据两个参数(log P和熔点(℃))来估算热力学溶解度。由于疏水性(溶解度有限的溶解度/疏水性:Kow)、结晶状态的稳定性(固态有限的溶解度/晶格能:Tm)或两者的原因,一种化合物可能表现出较差的溶解度。在过去十年中,一般溶解度方程中的每一个项都有所增加,这表明获批分子的溶解度已经下降。 随着时间的推移,FDA批准的分子的Mw也在增加(图1)。Sutherland等人对代表3773种化合物的173个独特的化学系列进行了审查,结果显示,Mw的增加对分子的溶解度几乎没有影响。解决水溶性差的两种最流行的处方方法是应用无定形分散体和脂质处方。最近对800种获批的口服分子进行的回顾性分析表明,在1940年至2017年期间,以脂质制剂或无定形分散体形式商业化的产品数量稳步增加。相比之下,打破晶格(无定形)以提高水溶性对高亲脂性分子来说可能是不够的,因为它们的吸收将受到溶解率的限制。这种经验性的共同变化确实清楚地表明,制药公司正在成功地将更复杂的制剂产品商业化,以弥补分子的溶解度和生物利用度的不足。 渗透性
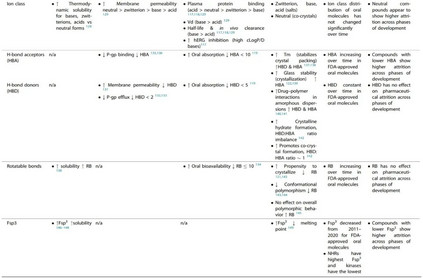
分子的极性是预测细胞渗透性的一个重要因素,极性大的分子一般渗透性小。使用PSA来预测小分子的渗透性,在文献中取得了不同程度的成功,许多作者强调了其优点和缺点。 根据Whitty等人的分析,要想获得口服生物利用性,Mw>700 Da的分子很可能显示出反复无常的性质,该分析指出,要想获得良好的水溶性,需要PSA≥0.2 Å2/Da×Mw,而3D PSA≤140 Å2对于良好的膜渗透性至关重要,但文献中相互矛盾的研究表明,PSA、Mw和溶解度之间不存在直接关联。 可旋转键 (rotatable bonds, RB) 的数量经常被用作分子灵活性的近似值和跟踪药物相似性的描述符。RB的平均数量与批准年份的关系图显示,随着时间的推移,更灵活的分子被批准的趋势更明显(图1)。分子的柔韧性可以提高溶解度(见Fsp3的讨论),但根据Veber及其同事的开创性分析,这限制了口服暴露。这是由于分子需要采取固定的构象与靶点结合,并通过膜,这涉及到熵的损失。 随着行业进入第四轮分子多特异性药物发现的浪潮,这类分子的运输特性预计将成为一个持续关注的领域,而且因为PSA被确定为限制某些类别的分子如基于PROTAC的多特异性药物的细胞渗透性的主要罪魁祸首。 分子的LogP(分配系数)是最常用的衡量亲油性的标准,并随着细胞渗透性的提高而呈正向趋势。除了在过去十年(2011-2020年)分子的平均cLogP(>1个对数单位)有统计学上的显著增加外,获批分子的cLogP通常被认为是评估亲油性的黄金标准,但它没有考虑到电离和H键对分配的影响,因为它只预测化合物的中性形式的分配。在化合物在生理条件下被电离的情况下,依赖pH值的cLogD可能是首选,它通常比电离分子的cLogP低。将分子描述符如Mw和亲油性(以logD衡量)结合起来是一种成功的方法,可以根据渗透性对分子进行分类和区分。 分子中HBA和HBD的数量可能是影响药物发现和CMC开发的最重要的分子描述符之一。随着时间的推移,FDA批准的口服分子的HBA数量增加(图1),因为药物中Mw、PSA和RB的数量增加。然而,HBD的数量并没有随着时间的推移而增加,这表明随着分子变得更加复杂和亲油,低数量的HBD是一种保留的特性。 一项研究对4176个安进公司化合物的结构特性进行了研究,结果表明,保持HBD数量(<2)和tPSA(<90 Å2)也会最大限度地提高逃避P-gp流出的概率。礼来公司的研究人员进一步指出,药物分入脂质膜和在膜内与P-gp结合是紧密耦合的过程,由于能量上不利的水解作用,分子中太多的H键基团会对渗透性产生负面影响,分子与P-gp腔的结合主要是由HBA驱动。从分子中去除HBD和HBA基团可能会对药效和靶点参与产生严重影响,因此,通过分子间和分子内的H键掩盖HBD和HBA可能是保持渗透性的一种可能策略。 ADMET特性
分子的口服吸收过程受诸如溶解度和渗透性等性质的制约,而这些性质又取决于分子的物理化学特性。一个分子的总体口服生物利用度是一个复杂的药代动力学参数,也包括清除成分。 在文献中,有许多帮助区分基于理化特性的ADMET特性的规则和指南的例子。其中被引用和讨论最多的是Lipinski’s rule of 5,该规则指出,如果一个分子违反了以下两个或两个以上的标准,就有可能出现口服吸收不良: Mw > 500 Da, clog P > 5, HBA > 10 和 HBD > 5 Veber等人在此基础上将RB<10和PSA<140 Å2作为提高口服生物利用度的考虑因素。这些理化性质中的大多数显示出随时间推移而增加的趋势(除HBD外,图1),表明它们对已批准药物的ADMET特性产生了越来越多的负面影响。 离子类别(由分子的pKa决定)是小分子药物的一个重要性质,直接影响到一系列ADMET结果和蛋白质-配体的相互作用。Martin等人调查了离子类别对口服生物利用度的影响,通常称为生物利用度评分(ABS)。 作者进一步提出,对于带负电荷的酸来说,重要的性质是PSA,而其他离子类的口服生物利用度可以通过Lipinski的五项规则来预测。随着时间的推移,FDA批准的口服分子的离子类分布没有明显变化(图2b),阴离子只占批准数量的20%∼。与其他离子类相比,带负电荷的酸类也有较高的血浆蛋白结合率、较低的分布容积和较低的体内清除率。血浆蛋白结合率也随着Mw和cLogP的增加而增加。碱基较高的分布体积导致半衰期的改善,但高cLogP/D的碱基显示出更多的hERG抑制、脱靶毒性风险和杂乱性。 体内清除率被认为是ADMET过程中最难预测的,因为它更依赖于化学结构而不是物理化学性质;离子化强烈影响清除率(由于血浆蛋白结合)。Mw对清除率没有影响,但随着cLogP的增加而增加。提高分子的亲和力和/或效力常常被推荐为补偿不良ADMET特性的一种方式。然而,在最近的一篇评论中,Leeson等人认为,更高的效力并不能弥补分子的不良ADMET特性(由于Mw和cLogP等物理化学特性的增加),因为随着时间的推移,获批药物的剂量和效力之间的相关性已经减弱了。**配体效率(LE)和亲脂配体效率(LLE)**是通常用来根据对靶点的亲和力对分子进行分流的指标,其中分子的效力被其大小(Mw/重原子数)归一化或被cLogP校正。作者进一步建议,从基于靶点的药物发现计划中出现的药物,其LE和LLE值最好有一个或两个大于所有已知作用于靶点的其他化合物的中位值。 可制造性
可制造性被定义为一种性能性质,它允许规模化生产和加工,同时控制成本并保证高质量,以满足安全和疗效的监管要求。 随着时间的推移,FDA批准的口服分子中RB的数量明显上升(图1)。一个RB被定义为任何单一的非环状键,连接到一个非末端的非氢原子上。较少RB的分子预计会更快地从溶液中结晶和沉淀出来。值得注意的是,RB(灵活性)和分子的整体多态性之间并不存在直接的关联。 HBA:HBD的比例也与水合物和溶解物的形成有关,在FDA批准的口服分子的数据中观察到的这一比例的增加可能表明溶解物形成的可能性增加。 从临床前给药的角度来看,pH值调整是一种强大的技术,通过它可以提高药物的溶解度和溶出度,但这种方法仅限于可电离的化合物、化合物的pKa和制剂的可接受的pH值范围。图2b显示,平均有70%的FDA批准的口服分子是可电离的。在过去的四五十年里,这一趋势似乎没有改变,大约有三分之二的分子是可电离的。这表明,pH值的调整仍然是分子递送的重要策略,相应地,盐的形成也是临床开发的有效策略。 高cLogP(>4)的分子一般适合使用基于脂质的给药系统,cLogP>5的分子可以利用淋巴系统来消除对生物利用度的首过效应。最后,过去十年中药物熔点的提高(基于Fsp3的降低和HBA的提高)也表明,它们应该更适合采用自上而下的粒度减小技术来提高溶出率,并在某些情况下改变药物的表观溶解度(纳米化),因为高熔点药物在高能自上而下的粒度减小过程中对晶体形态的改变更有抵抗力。 制药损耗的趋势
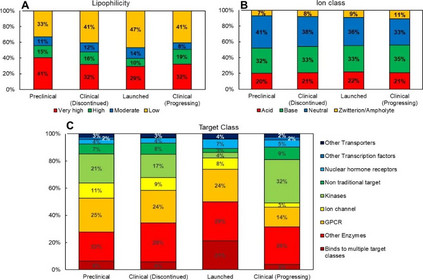
众多研究表明,制药损耗的增加是药物理化性质不理想的直接后果。 临床试验I期的损耗通常与分子的安全性和耐受性有关,而II期及以后的损耗则是由于药物的疗效和调节疾病状态下的靶点的能力。有许多研究将分子的毒性结论与分子的亲脂性直接联系起来,最引人注目的是辉瑞公司的3/75法则,而效力则通过分子大小、亲脂性、H键和三维结构与配体效率指标联系起来。 随着时间的推移,FDA批准的口服分子有一个明显的Mw和RBs增加的趋势(图1),但在这里提出的数据中,没有迹象表明这些性质中的任何一个会影响化合物的损耗,这可以从推出的、临床(活性或非活性)和临床前分子之间的相对大小差异看出(图4)。PSA、Fsp3和HBA数值越高表示成功的可能性越大,而cLogP和cLogDpH6.5数值越低表示成功的可能性越大。这五个性质的趋势在统计学上是显著的,P值<0.05。 cLogP和cLogDpH6.5的影响大小最为突出,临床前和上市分子之间的分配系数几乎下降了一个pH单位。cLogP和cLogD pH6.5的下降进一步补充了PSA的适度增加,表明极性的、亲脂性较低的分子有更大的成功可能性。然而,目前正在进行临床试验的分子的cLogP、cLogD pH6.5和PSA值明显高于上市的分子。 总的来说,较高的分配系数和较低的ePSA表明,目前在临床上进展的分子比上市的分子更具亲油性(图5a)。这种亲脂性的趋势预计会继续下去,因为随着时间的推移,靶点变得越来越复杂,特定的靶点家族(NHRs)被追求,保持(和提高)效力仍然是一个关键的目标。 已上市分子的平均Fsp3为0.45,接近Lovering等人提出的0.47的数值,在这个数值以上的分子显示出更高的临床成功可能性;但是,目前正在进行临床开发的分子,这个数值下降到0.37。造成这种下降的原因之一是目前处于临床开发阶段的激酶靶向分子大幅增加(32%),而已上市(4%)和已停止临床开发(17%)的分子则大幅增加(图5c)。如下文所述,由于嘌呤环的普遍存在,这些药物的Fsp3较低。离子类别似乎对化合物的流失没有重大影响;但是,带电分子(酸、碱和齐聚物的总和)与中性分子相比,似乎有更大的成功可能性(图5b)。
图4. 理化性质与发展阶段的关系
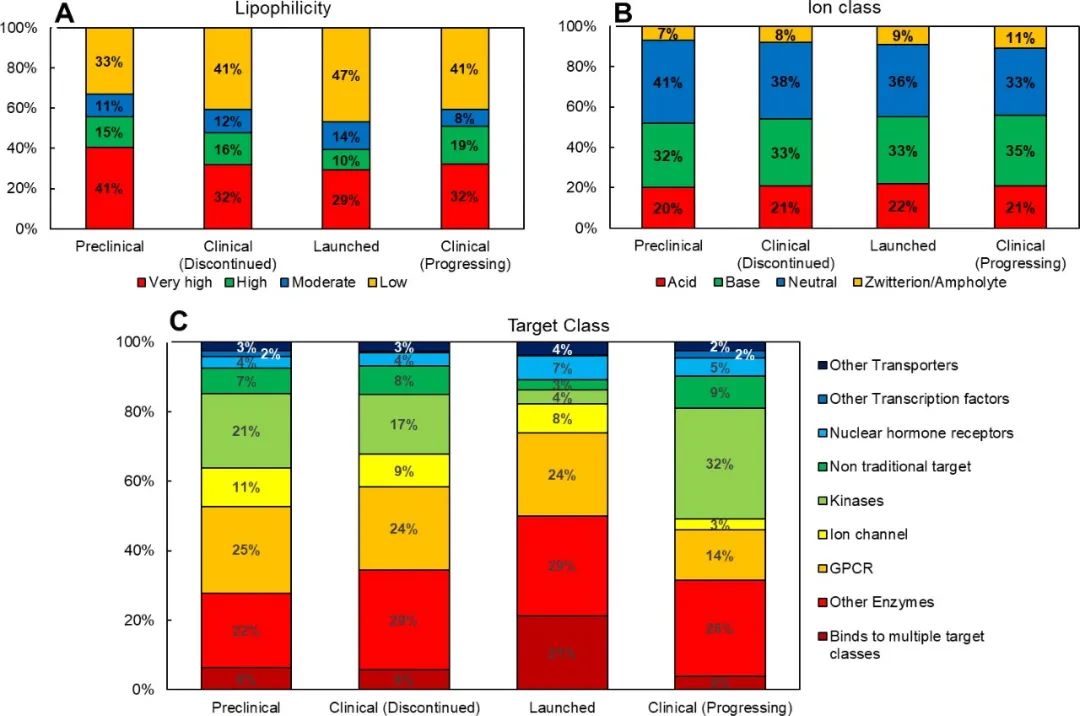
图5. 分子亲油性(a)和离子类(b)及靶点类别(c)的柱状图与开发阶段的关系 损耗分析的目的是确定与理化性质有关的趋势和风险。 例如,在过去十年(2011-2020年),FDA批准的口服分子的分配系数(cLogP和cLogDpH6.5)有所增加(图1),而损耗分析表明,分配系数较低的分子更有可能上市(图4c)。21世纪制药业的现实之一是减少开发时间和接触病人的压力,这就产生了一种”快速失败和快速学习”的心态。因此,第一代分子可以迅速进入临床,以测试治疗假设,因为他们知道,具有改进特性的、符合最新候选靶点的分子(第二代分子)可以迅速跟进。这类第一代或first-in-class分子预计会在”活跃的临床开发”数据集中占比过高,因为2010-2019年批准的新药中有37%是first-in-class疗法,比2008年和2009年的17%和29%都要高,这可能是目前正在开发的分子中物理化学性质提高的一个原因。 理化性质与药物靶点类别的关系
新兴的基于靶点类别的药物发现范式表明,在蛋白质家族中获得的科学专长和知识可以降低发现项目的技术失败。
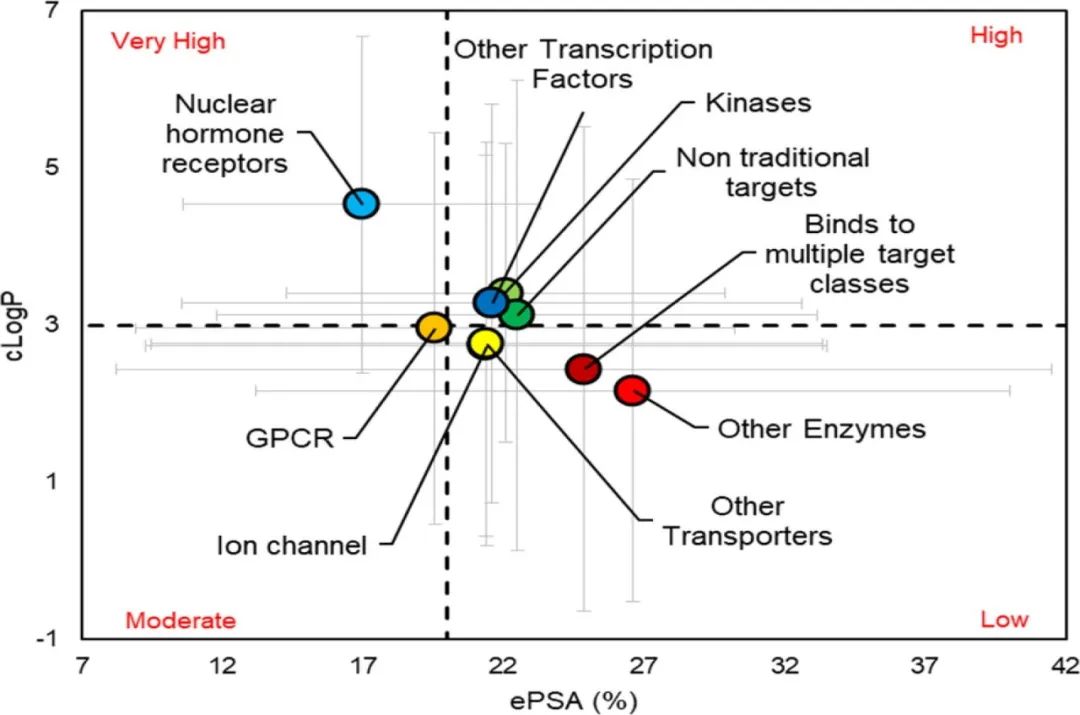
图6. 基于靶点类别的ePSA(有效极性表面积)与cLogP(计算的辛醇-水分配系数)的散点图
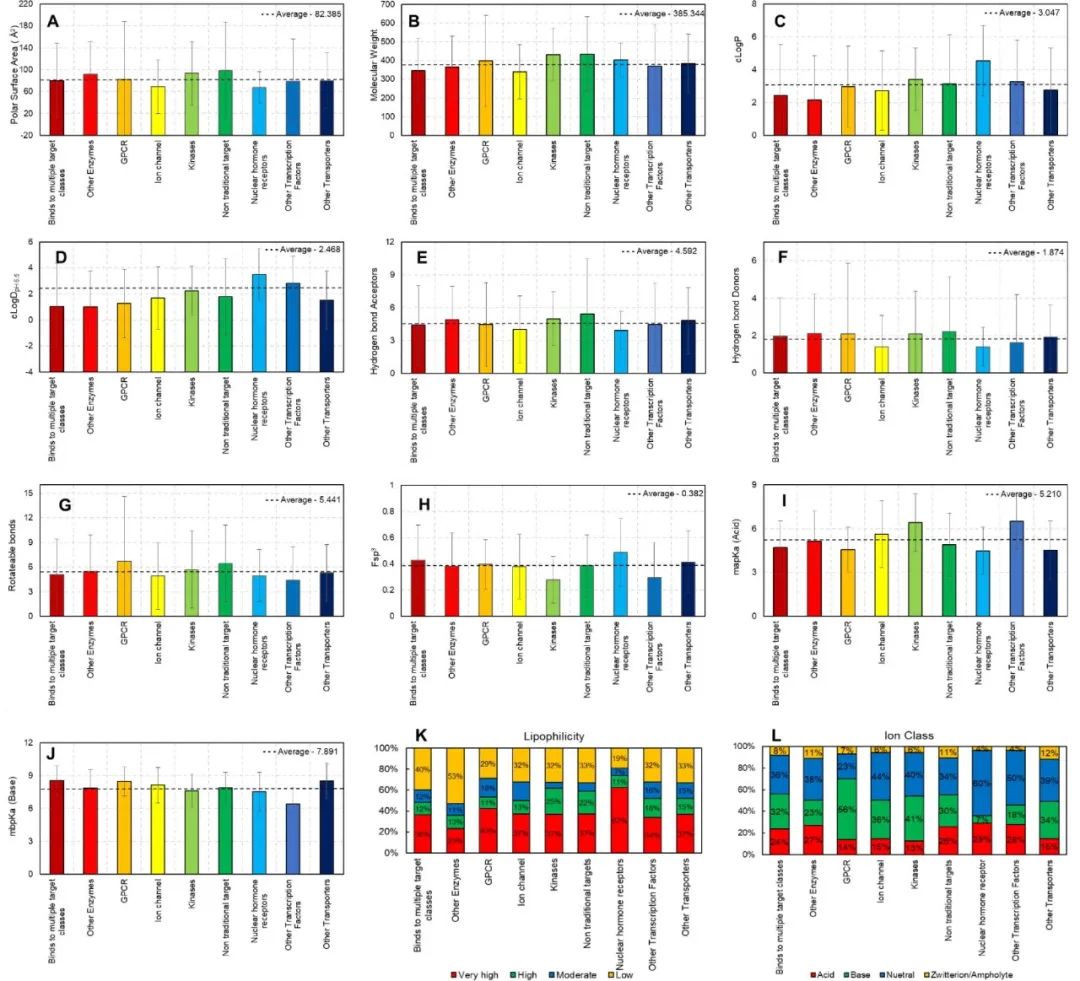
图7. 理化性质(PSA(a),Mw(b),cLogP(c),cLogDpH6.5(d),HBA(e),HBD(f),RB(g),Fsp3(h),酸的最大pKa(mapKa)(i),碱的最大pKa(mbpKa)(j),亲脂性(k)和离子类别(l))与靶点类别关系的条形图 在目前的数据集中,与非传统靶点结合的分子似乎具有最高的PSA(图7a);然而,它们的高Mw(图7b)抵消了极性的整体增益,减少了ePSA(图6)。与非激酶(其他酶)结合的分子似乎比与其他靶标类结合的分子具有更高的ePSA(在P < 0.05时有统计学意义)。而与NHRs结合的分子具有最低的PSA和ePSA(在P<0.05时有统计学意义)。 与NHRs结合的分子需要穿过质膜来调控它们的靶点,这与通过膜受体作用的分子不同。因此,这些分子比那些与膜锚定和膜驻留的酶结合的分子更亲油,极性更小,包括大类的GPCRs、离子通道和运输工具。 有趣的是,一些家族间的差异显现出来。与NHRs结合的分子似乎与针对其他转录因子的分子有非常不同的理化性质,例如STAT抑制剂(图6,图7)。同样,与激酶结合的分子比与其他酶(非激酶)结合的分子更具亲油性,这是因为针对激酶的分子有更多的芳香环。 对特定靶点蛋白类别的离子类别的分析比较表明,60%的与NHRs结合的分子是中性的,而其余的大部分(29%)是酸性的(图7i),这表明在大多数情况下,处理NHRs的团队将无法获得可电离的碱性pKa来提高这些分子的水溶性。 对于与激酶结合的分子,高的cLogP、HBA和HBD,以及较低的Fsp3(表明高Tm),表明这两个因素(疏水性和晶格能)导致其溶解度差(图7)。靶向GPCRs的分子主要是含碱的化合物(56%),高于任何其他靶点类别(图7i)。这可以解释为A类胺基GPCRs中保守的天冬氨酸残基对其结合配体所含的碱性氨基的识别。 结束语和未来展望
从DevMA的角度来看,就靶点类别而言,以激素受体为靶点的分子是最亲脂的,以脂质制剂的形式提供它们可以预期是一种合理的策略。而与激酶结合的分子溶解度较差,这是由于与目标物结合所需的疏水性(由于芳香性)、较低的sp3碳比例以及相对较高的HBA和/或HBD数量导致分子具有高晶格能(熔点)。 与GPCR结合的分子应该是阳离子和柔性的。HBD是唯一保持不变的特性,这表明它是整个行业和长期以来被化学家保留的一个重要特性。Mw、RB、HBA和PSA的趋势在几十年间呈现出统一的增长。 对药物再利用中心数据集的药品损耗分析表明,cLogP、cLogD、HBA、PSA和Fsp3是损耗的指标,而Mw、RB和HBD则没有影响。 参考资料 Agarwal P, Huckle J, Newman J, Reid DL. Trends in small molecule drug properties: A developability molecule assessment perspective. Drug Discov Today. 2022 Sep 17;27(12):103366. doi: 10.1016/j.drudis.2022.103366. Epub ahead of print.
--------- End ---------