传统的药物开发是一个困难、昂贵且耗时的过程。平均而言,将新药推向市场需要10~15年和15亿~26亿美元。药物重定位旨在发现现有或废弃药物的新用途,这些药物以前通常经历过Ⅰ期临床试验、满足特定安全要求以及通常已经获得人类使用许可,从而为药物开发提供了一种更快速、风险更低、成本更低的有效替代方法。最近发表的综述文章”Recent computational drug repositioning strategies against SARS-CoV-2”,就以SARS-CoV-2为例,总结了药物重定位的可用数据集与不同计算策略等,可以为准备进行相关研究的人员或者药物发现新入门者等提供一定的参考信息。 ——可用数据**——**
随着越来越多相关研究的开展,新冠病毒的数据逐渐积累,美国国家生物技术信息中心(NCBI)收集和总结了新冠病毒基因组、转录组学和蛋白质组学表达谱、蛋白质结构、病毒-宿主蛋白相互作用以及与其他冠状病毒变异相关关系等一系列数据,资源可在网站:https://www.ncbi.nlm.nih.gov/sars-cov-2/ 获得。而现有药物的相关知识主要包括化学结构、药物靶标相互作用、药物干扰、表型效应和药物分类等,这些数据可以从ChEMBL、PubChem、LINCS等数据库获得,表1给出了一些可用数据库的分类描述及网址。
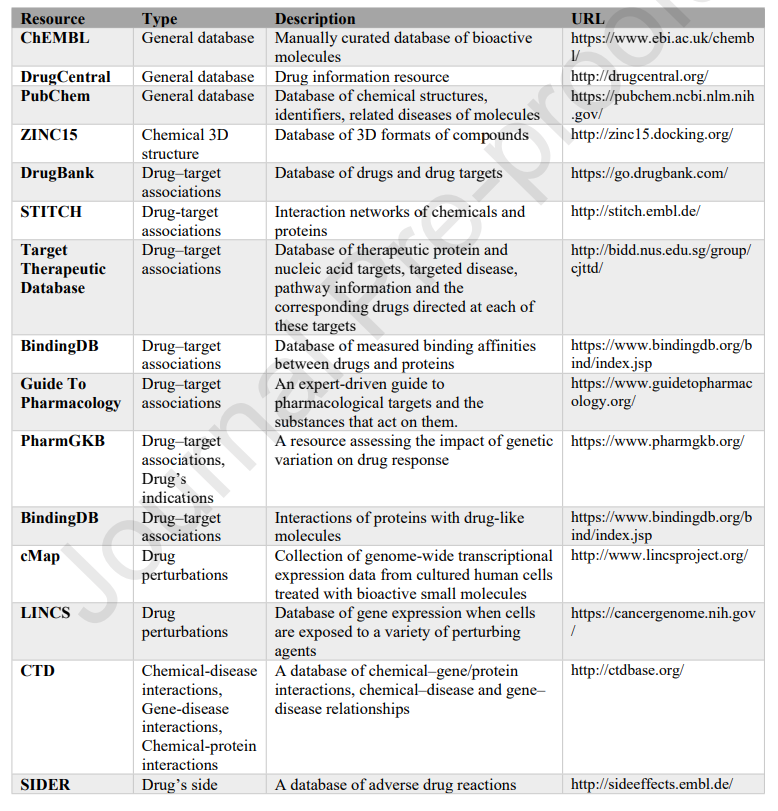
表1. 药物相关数据库分类及描述**——药物重定位的计算策略****——**前面所述的病毒和药物所有数据可主要分成四种类型:序列数据、结构数据、表达数据和相互作用数据。对于不同的数据类型,可使用不同的方法来重定位药物,如图1所示。
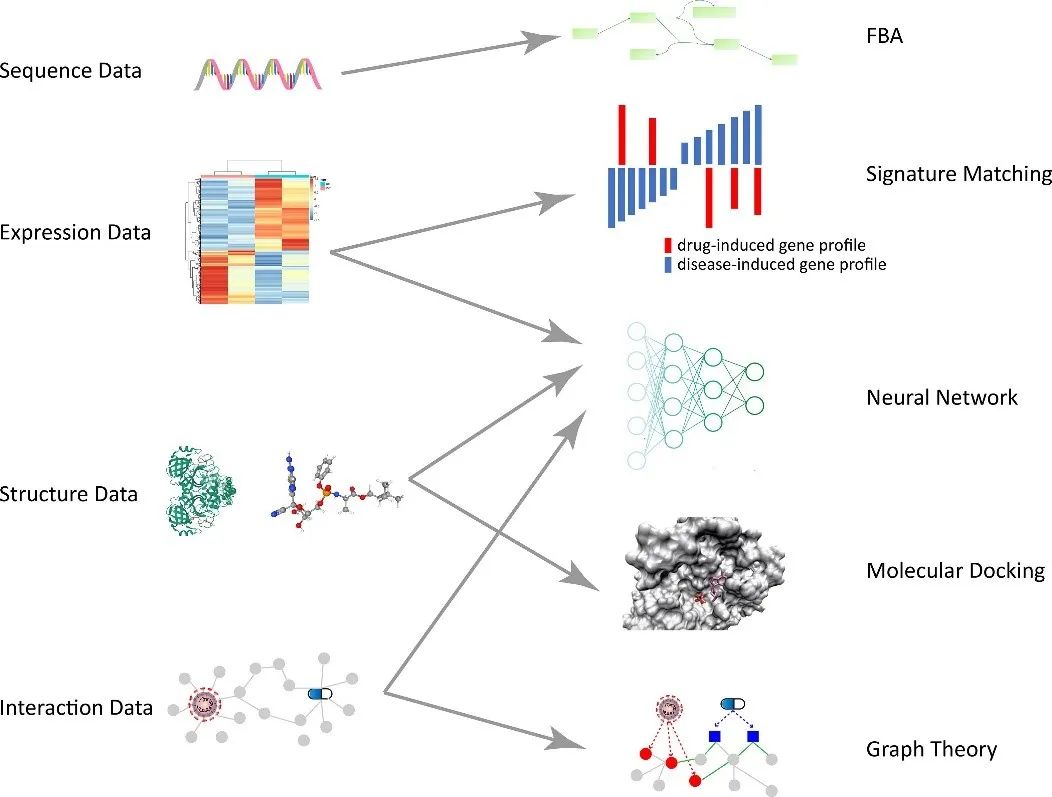
图1. 可用数据与相应方法之间的关系。FBA即流平衡分析(flux balance analysis),一种可以用来构建和模拟分析基因组级别代谢网络的数学方法 一、基于序列的计算策略基因组序列是病毒遗传的基础,病毒核苷酸序列可以掺入由节点(代表化合物或代谢物)和边(代表由一种或多种酶催化的反应)组成的代谢网络中。在此表示中,由于特定酶而积累的化合物浓度过高可导致观察到的特定病理现象,这些酶就可以被考虑用作治疗特定靶标。流平衡分析(FBA)经常作为分析这种代谢网络中代谢物流动的方法,流就是代谢网络中化学反应的反应速率,在稳态下,反应速率满足一定的分布(流分布)。FBA即在限制条件下求目标函数的最优解,得到流分布和相应的目标值。 比如Renz等人将基因组和蛋白质序列转化为核苷酸和氨基酸的技术,然后将这些核苷酸、氨基酸、三磷酸腺苷和SARS-CoV-2的焦磷酸释放整合到受感染的肺泡巨噬细胞模型中,再构建重要生物量的目标函数,通过优化目标函数的增长率,他们发现了未感染和受感染宿主细胞之间的化学计量和代谢变化,最终确定鸟苷酸激酶或许可用于SARS-CoV-2的抗病毒治疗。 二、基于基因表达的计算策略
使用药物治疗干预时需要考虑疾病系统属性的扰动,这与遗传和基因组本身关系不大,而可以用基因表达变化来描述。COVID-19的表达谱可以从GEO或Array Express数据库检索到,其中包含了人类和动物模型中数百种疾病状况的原始基因表达数据。同时,药物扰动数据可以从Connectivity Map(cMap)数据库得到,它包括通过在五种人类癌症细胞系中投加1300种化合物产生的基因表达谱(GEP)。cMap的下一代是L1000平台,它包含了用2万个化合物处理50个人类细胞系后产生的140万个GEP。基于这些基因表达数据,特征匹配和神经网络的方法可用于药物重定位。 特征匹配
特征匹配,也被称为特征逆转,其涉及调查疾病表达模式是否可以在分子水平上被逆转,这已被专门应用于COVID-19,并被证明具有潜在的治疗益处。一方面,SARS-CoV-2的特征可以通过比较新冠病毒患者和未受影响的对照组之间的基因表达谱来确定。另一方面,可以通过用小分子药物治疗前后的表达变化来评估分子特征。一个常用的特征匹配方法是基因富集分析(GSEA),这是一种确定先验定义的基因集在两个生物状态之间是否存在统计学显著差异的计算方法。在我们的情景中,GSEA用于确定新冠病毒患者和未受影响对照组之间的差异表达基因是否在药物扰动表达谱中富集。****
神经网络
神经网络近年来发展迅速,促进了自然语言处理和图像识别等方面的成就。基于表达数据,它们已被用于学习新冠病毒和药物基因图谱的嵌入表示,然后计算两者图谱的相关性评分,与新冠病毒最负相关的化合物被视为治疗候选药物。
三、基于结构的计算策略
结构是功能的基础,SARS-CoV-2相关蛋白质和化合物的结构也可用于药物重定位。蛋白质结构信息包括氨基酸序列、二级结构和3D结构。化合物分子除了2D结构和3D构象,还具有线性表示如SMILES、inChi等。 神经网络
对于一维结构数据,核苷酸序列、氨基酸序列和分子的SMILES等类似于自然语言,可以用人工智能/深度学习的方法来预测药物分子-蛋白质关联。分子对接
如果可以获得与新冠病毒和药物相关的3D构象,则可以使用分子对接来模拟它们的相互作用,预测药物靶标复合物的结合构象及结合能。相关研究主要分为三类:1、针对SARS-CoV-2蛋白的分子对接,通常是靶向主蛋白酶或RNA依赖性RNA聚合酶等靶标;2、针对宿主蛋白如跨膜丝氨酸蛋白酶2等的分子对接;3、同时考虑SARS-CoV-2蛋白和宿主蛋白的对接。 四、基于相互作用的计算策略
基于结构的策略对药物和蛋白质进行了建模,但单个蛋白质的运作并不是孤立于复杂系统的,还包含了许多其他蛋白质的参与。因此,每个药物与靶标的相互作用都需要在其综合背景下进行研究。科学家们已经通过实验或计算机的方法确定了病毒蛋白和宿主蛋白之间的许多相互作用,并将这些病毒-人类相互作用于人类蛋白质-蛋白质相互作用和药物-靶标相互作用相结合,构建成一个网络 ,网络的节点代表各种生物实体(药物、疾病或蛋白质),网络的边代表相互作用。图论分析和神经网络的方法都可以系统地探索整个生物网络,图论分析利用网络邻近性来寻找靶向疾病模块的药物,神经网络则可以用来预测药物和靶标之间的缺失环节。 **——小结——**1、上述的方法根据不同类型数据进行药物重定位,但每种类型数据仅代表生物系统的部分愿景,整合不同层次的数据可以进一步提高药物重定位的成功率。2、虽然由于使用的数据类型不同,每种方法无法直接比较,以及重定位后的药物依然需要验证实验,但总体而言,药物重定位的计算方法可以为流行病应对提供快速有效的重要指导。 参考文献:
L. Lu et al., Recent computational drug repositioning strategies against SARS-CoV-2. Computational and Structural Biotechnology Journal (2022). DOI: 10.1016/j.csbj.2022.10.017.
点击左下角的"阅读原文"即可查看原文章。
作者:王丽莹 审稿:黄志贤 编辑:王丽莹
GoDesign ID:Molecular_Design_Lab ( 扫描下方二维码可以订阅哦!)

本文为GoDesign原创编译,如需转载,请在公众号后台留言。