2022年9月24日,浙江大学药学院侯廷军教授团队在Drug Discovery Today上发表文章”Machine learning methods for pKa prediction of small molecules: Advances and challenges“。论文中作者总结了用于预测pKa的基于机器学习的两类QSAR模型(基于描述符的模型和基于图的模型),并对pKa预测当前遇到的挑战和未来的方向进行了深入的讨论和分析。

1 摘要pKa是影响小分子的许多ADMET性质的基本属性。然而,快速且准确的pKa预测仍然是一个巨大的挑战。在这篇综述中,作者概述了基于机器学习的QSAR模型预测pKa的当前进展,包括基于描述符的方法和基于图的方法,并总结了它们的优缺点。此外,作者还从实验数据、影响pKa的关键因素和计算预测工具三个方面强调了当前遇到的挑战和未来的研究方向。
2 介绍
pKa是一个基本的物理化学参数,广泛应用于药物化学、有机合成、生物化学、环境科学和材料科学。在药物发现中,pKa决定了类药分子在不同pH范围的特定组织和器官下的主要质子化形式,因此对其生物活性、ADMET等性质有很大影响。 作为实验技术的一个不可缺少的补充,用计算方法预测pKa更有效,也更便宜。现有的pKa预测方法可分为两类:经验方法和基于物理的方法。经验方法包括线性自由能关系(linear free energy relationship, LFER)和QSAR模型。基于物理的pKa预测依赖于反应自由能计算,通常结合线性经验修正(linear empirical corrections, LECs)以吸收系统误差。 在过去的十年中,基于机器学习(ML)技术的QSAR建模在pKa预测方面取得了显著的成功。具体来说,QSAR方法可以分为(图1):
- 基于描述符的模型 (descriptor-based models),
- 基于图的模型 (graph-based models)。
基于描述符的模型以专家提取的固定维特征向量作为输入。相比之下,基于图的模型直接作用于由基本原子和键属性注释的分子图。这篇综述概述了基于ML的小分子pKa预测的最新进展,总结了计算预测工具,并讨论了当前的挑战和未来的方向。
图1 基于机器学习的QSAR预测pKa概览。紫色和绿色虚线框分别表示基于描述符的模型和基于图的模型。缩写:SVM,支持向量机;ANN,人工神经网络;ISOAK,迭代相似最优分配核;GNN,图神经网络。
3 基于描述符的模型
基于描述符的模型的预测精度取决于输入特征。目前,有数千种定量描述符和定性分子指纹可用于表示小分子。对于pKa预测,原子描述符(atomic descriptors)和根指纹(rooted fingerprints)被广泛用于表示电离中心周围的局部环境。这些局部表示可以自然地应用于预测微pKa (micro-pKa)。 3.1 原子描述符
本文中,原子描述符是指描述某个原子或化学键的实值参数。从量子力学(quantum mechanics, QM)中导出的大多数原子描述符可以通过从头计算、密度泛函理论(density functional theory, DFT)或半经验计算获得。为了快速估计,它们也可以通过经验方法获得,如鲍林电负性(Pauling’s electronegativity)和盖斯泰格部分电荷(Gasteiger partial charge)。现有工作验证了pKa值与各种原子描述符之间的关系,包括partial atomic charges、Fukui frontier molecular orbital (FMO) 描述符、基团亲性指数(group philicity index)、量子拓扑分子相似性(quantum topological molecular similarity, QTMS)描述符,密度泛函反应理论(density functional reactivity theory, DFRT)描述符等。作者在文中回顾并总结了相关方法,分析了它们的优缺点以及当前的挑战。
3.2 根指纹
根指纹是基于二进制或计数的特征向量,用于描述指定根原子周围的局部结构环境。这里,根原子是指电离原子。作者在文中回顾并总结了大量基于根指纹的pKa预测方法。
3.3 混合特征
近年来,研究人员对分子特征和ML算法的各种组合进行了广泛的实验,并在开源数据、代码和预测工具方面做出了重大贡献。混合特征(Hybrid features),即描述符和分子指纹,可以相辅相成,因为描述符强调物理化学性质,而指纹侧重于结构信息。论文中作者也总结了同时考虑两类特征的多个具有代表性的模型和方法。
4 基于图的模型
小分子可以自然地描述为图,其中节点和边分别表示原子和化学键。基于图的方法可以直接从注释的分子图中提取信息,在分子性质预测方面具有很大的优势。
4.1 图核
传统上,图结构数据可以用图核(graph kernels)来表示,图核在图上计算内积来度量它们的相似性。图核允许核化机器学习方法直接处理图,而无需从图到特征向量的中间转换,从而避免了结构信息的丢失。值得注意的是,图核方法在具有高度结构多样性的大量化合物上表现得更好,显示了其开发通用模型的潜力。
4.2 图神经网络
尽管图核可以直接操作图形,但它们仍然是手动设计的,无法学习为下游任务定制最佳表示。在这种背景下,科学家将卷积网络(CNN)扩展到图结构化数据上,并提出图神经网络(graph neural networks, GNN)模型,例如,图卷积网络(graph convolutional networks, GCN)、消息传递神经网络(message passing neural networks, MPNN)、图注意力网络(graph attention networks, GAT)、有向MPNN (directed MPNN)、Attentive FP (Attentive fingerprints)等模型。研究者也基于GNN提出了大量的pKa预测模型,如图2所示。
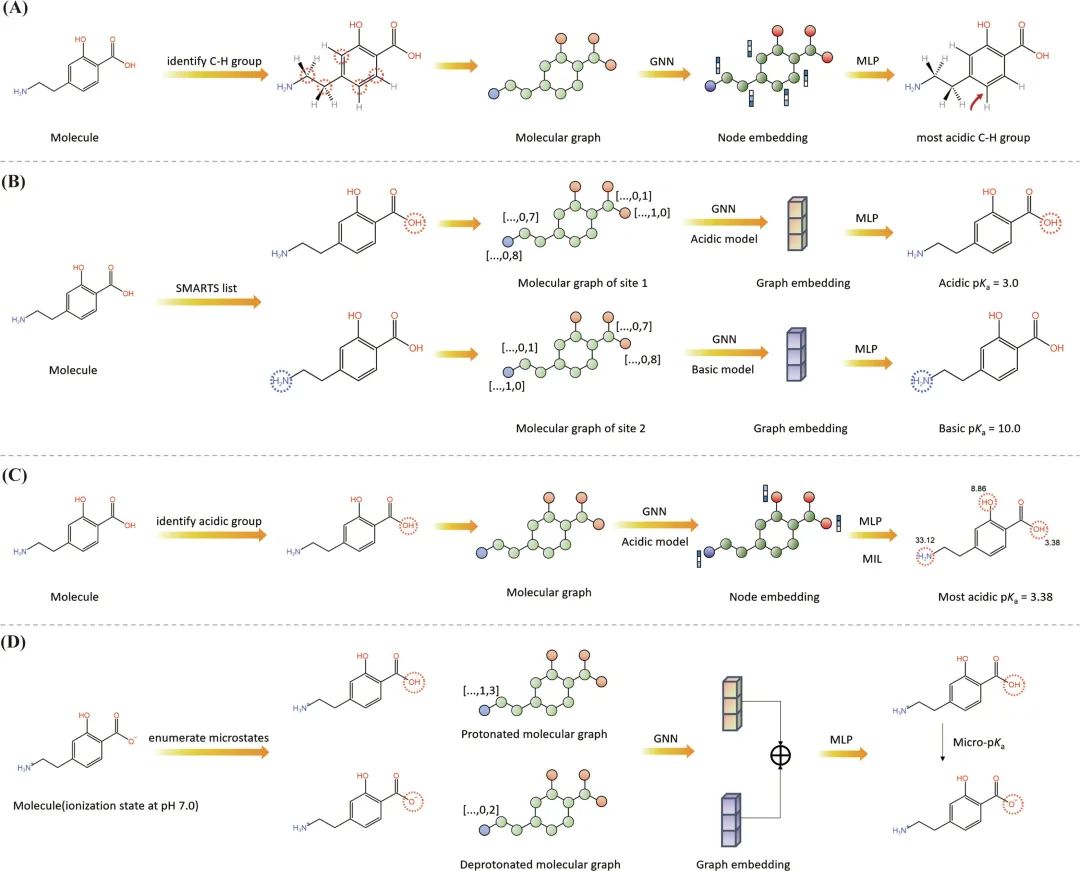
图2 基于GNN的pKa预测模型的图示。(a) Roszak的模型。(b) MolGpKa:原子特征是指电离中心标志和到目标中心的最短距离。(c) Graph-pKa。(d) pKasolver:原子特征是指形式电荷和氢原子的总数。红色和蓝色圆圈分别表示酸性和碱性电离中心。缩写:GNN,指图神经网络;MLP,指多层感知器。
5 挑战和未来方向
由于数据稀缺性和特征的内在复杂性,精确预测pKa仍然是一个具有挑战性的问题。目前,开源数据的数量和质量都比较欠缺。 另一个挑战是复杂因素影响特定基团的电离,包括灵活的构象、对称的结构、罕见的杂环、多电离中心、共轭体系中的电荷转移、互变异构和分子内或分子间相互作用。一个潜在的解决方案是将领域知识与最先进的深度学习(deep learning, DL)算法(如3D GNN)结合起来,以更好地从训练数据中捕获化学模式。 表1总结了预测pKa的基于QSAR的计算工具。从中,可以得出以下结论:
- 越来越多的开源预测工具正在涌现;
- 数据驱动特征正在逐渐取代人工提取的特征;
更多的注意力正在从建立类特异的模型转向建立通用模型。
与商业软件相比,开源工具仍然存在重大不足。首先,他们无法全面解决三个与pKa相关的任务,即微观pKa预测、宏观pKa(macro-pKa)预测以及不同pH条件下每个微观状态的比例。其次,自动化程度有待提高,主要体现在互变异构体的枚举、多步pKa预测和批量评估。最后,研究者对模型的可解释性研究较少,尽管这是使预测合理化以说服化学科学家的一个重要步骤。此外,它可能有助于发现以前未知的化学知识并指导先导化合物的优化。 表1 用于pKa预测的基于QSAR的计算工具
6 结论
pKa的计算预测对化学科学,特别是药物发现有着深远的影响。在过去的几十年里,研究人员在pKa预测领域取得了显著进展,并开发了各种可访问的工具。然而,仍有多个问题有待解决,主要原因是数据不足和结构因素产生的复杂影响。 众所周知,基于QSAR模型的预测能力在很大程度上取决于输入特征的质量。幸运的是,GNN的出现为直接从分子图中学习具有表现力的特征提供了一种智能解决方案。GNN在pKa预测任务中的优势,无论是微观还是宏观的pKa,在最近的研究中都得到了很好的证实。作者认为,利用专家知识和GNN架构的协同作用,有可能从较少的数据中捕获更多的结构-pKa关系,从而克服上述瓶颈,构建更可靠的模型。 全面评估现有方法的预测能力和应用领域,有利于模型的选择和改进。作者认为,高质量数据的积累和强大算法的出现将有助于开发准确、高效、通用和可解释的pKa预测模型。关于未来的应用,pKa预测工具可以作为人工智能(AI)驱动药物发现工作流程中的插件;例如,分子生成中的性能优化和虚拟筛选中的性能过滤。因此,科学界提出的模型可以在实践中发挥作用,切实降低失败风险。参考资料
Jialu Wu, Yu Kang, Peichen Pan, Tingjun Hou, Machine learning methods for pKa prediction of small molecules: Advances and challenges, Drug Discovery Today, Volume 27, Issue 12, 2022, https://doi.org/10.1016/j.drudis.2022.103372. --------- End ---------