药物发现和开发对制药业和患者具有巨大的潜在利益。预测药物-靶标亲和力 (DTA) 有利于加速药物发现。图神经网络 (GNN) 已广泛用于 DTA 预测。然而,现有的浅层 GNN 不足以捕捉化合物的全局结构。此外,基于图的 DTA 模型的可解释性高度依赖于图注意力机制,无法揭示分子中每个原子之间的全局关系。
近日,中山大学智能工程学院陈语谦教授团队在国际知名杂志Chemical Science 在线发表了题为“MGraphDTA: deep multiscale graph neural network for explainable drug–target binding affinity prediction”的研究论文。该研究提出提出了一种基于化学直觉的深度多尺度图神经网络用于 DTA 预测 (MGraphDTA)。在 GNN 中引入了密集连接,并构建了一个具有 27 个图卷积层的超深度 GNN,以同时捕获化合物的局部和全局结构。他们还开发了一种新颖的视觉解释方法,梯度加权亲和激活映射 (Grad-AAM),从化学角度分析深度学习模型。作者使用七个基准数据集评估了他们的方法,并将所提出的方法与最先进的深度学习 (DL) 模型进行了比较。MGraphDTA 在各种数据集上显着优于其他基于 DL 的方法。此外,结果表明 Grad-AAM 创造了与药理学家一致的解释,这可能有助于我们直接从人类感知之外的数据中获得化学见解。这些优点表明,所提出的方法提高了 DTA 预测建模的泛化和解释能力。

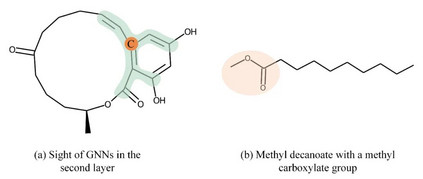
1 研究背景 药物-靶标亲和力(DTA)预测有利于加速药物筛选的过程。现有的高通量筛选实验用于确定药物和靶标之间的生物活性是一个昂贵费时的过程。因此,基于已经在临床实验中测量的药物-靶标亲和力,使用统计学和机器学习模型来估计新的药物-靶标的亲和力是重要的替代方案。由于化合物的信息可以用图来描述,所以图神经网络(GNNs)已经被广泛应用于DTA预测。为了提高DTA预测的精度,图神经网络的设计必须考虑如下三点:第一,为了捕捉到化合物的全局结构,GNNs的深度要足够深;第二,GNNs要具备保留局部结构的能力;第三,所设计的GNNs模型要具备可解释性。然而,目前基于浅层GNNs的DTA预测方法既无法捕捉化合物的全局结构,也无法很好地保留化合物的局部结构。例如,两层的GNN的感受野无法覆盖到化合物的环状结构(图1(a)),也无法很好地保留小的局部结构(图1(b))。
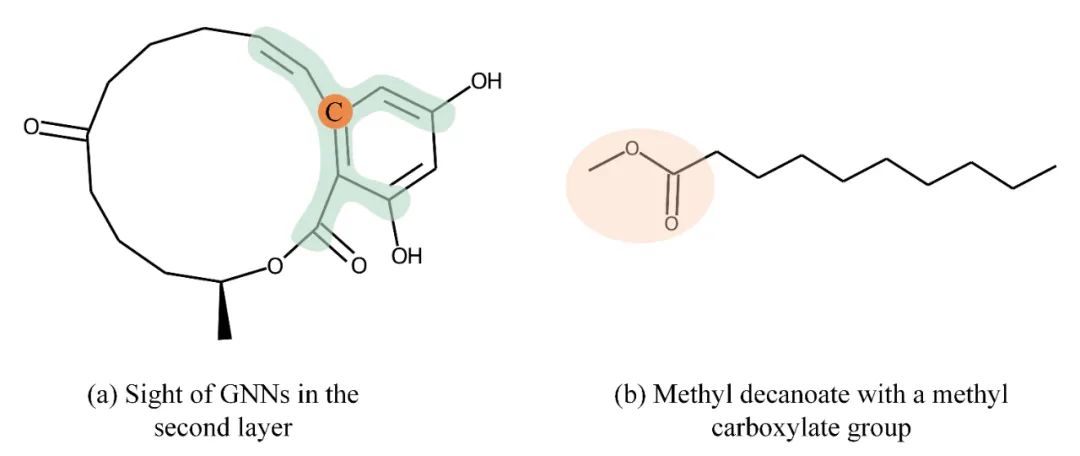
图1. 图神经网络的设计要点
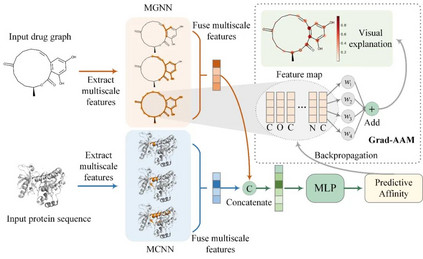
2 文章简介 针对上述问题,中山大学智能工程学院智能医疗中心陈语谦教授团队报道了一种基于深层多尺度图神经网络预测药物-靶标亲和力的方法MGraphDTA。MGraphDTA分别通过多尺度图神经网络(MGNN)和多尺度卷积神经网络(MCNN)来提取药物和靶标的特征,并结合这两种特征来预测亲和力(图2)。其中,MGNN通过引入稠密连接来拓展网络的深度和保留局部特征,从而使模型能够同时捕捉到化合物的全局结构和局部结构。相关研究成果发表在Chemical Science上。
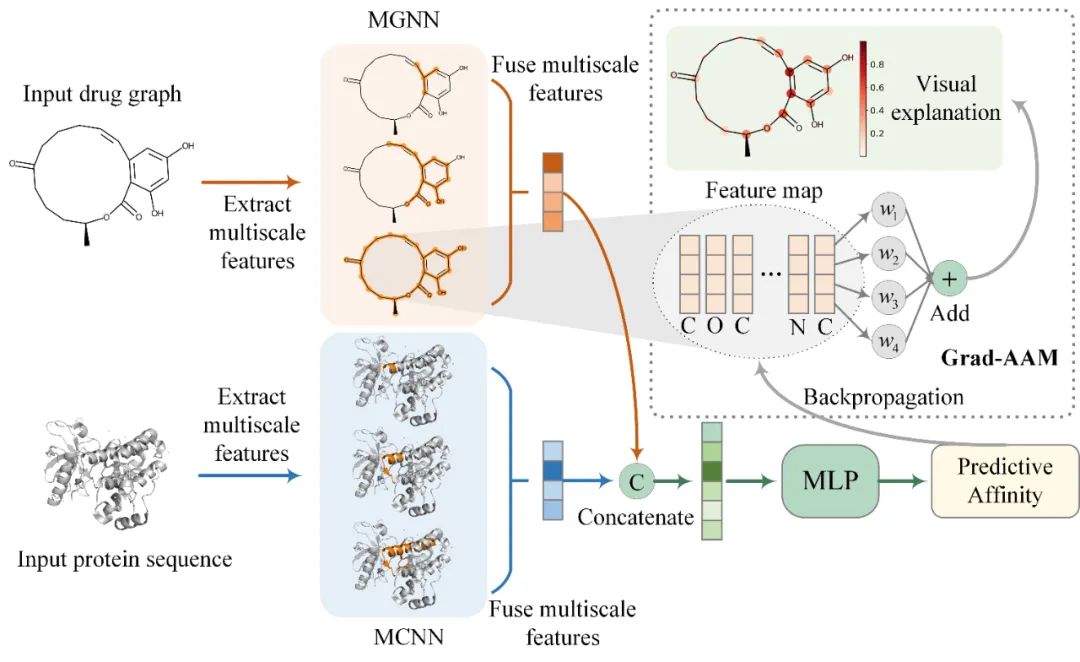
图 2. MGraphDTA的总体框架
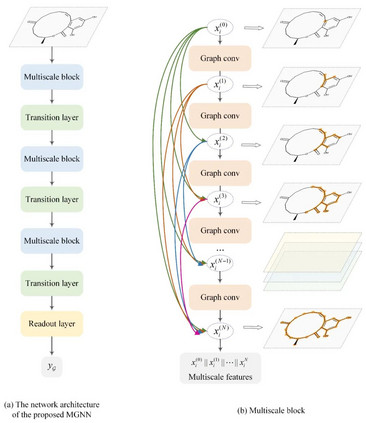
3 研究内容 基于多尺度图神经网络(MGNN)的药物特征提取 GNNs的感受野受网络深度影响。一般而言,网络越深,其感受野越大,GNNs能感知的化合物亚结构越大。针对已有的浅层GNNs无法捕捉化合物的全局结构的问题,MGNN引入了稠密连接来拓展网络的深度和保留局部特征(图3)。稠密连接可以有效地缓解梯度消失问题,从而拓展了模型的深度。通过稠密连接,不同感受野的特征图被拼接在一起,从而形成多尺度特征并作为下一个图卷积模块的输入。由于多尺度特征既包含全局特征又包含局部特征,所以MGNN能够同时捕捉到化合物的全局结构和局部结构。
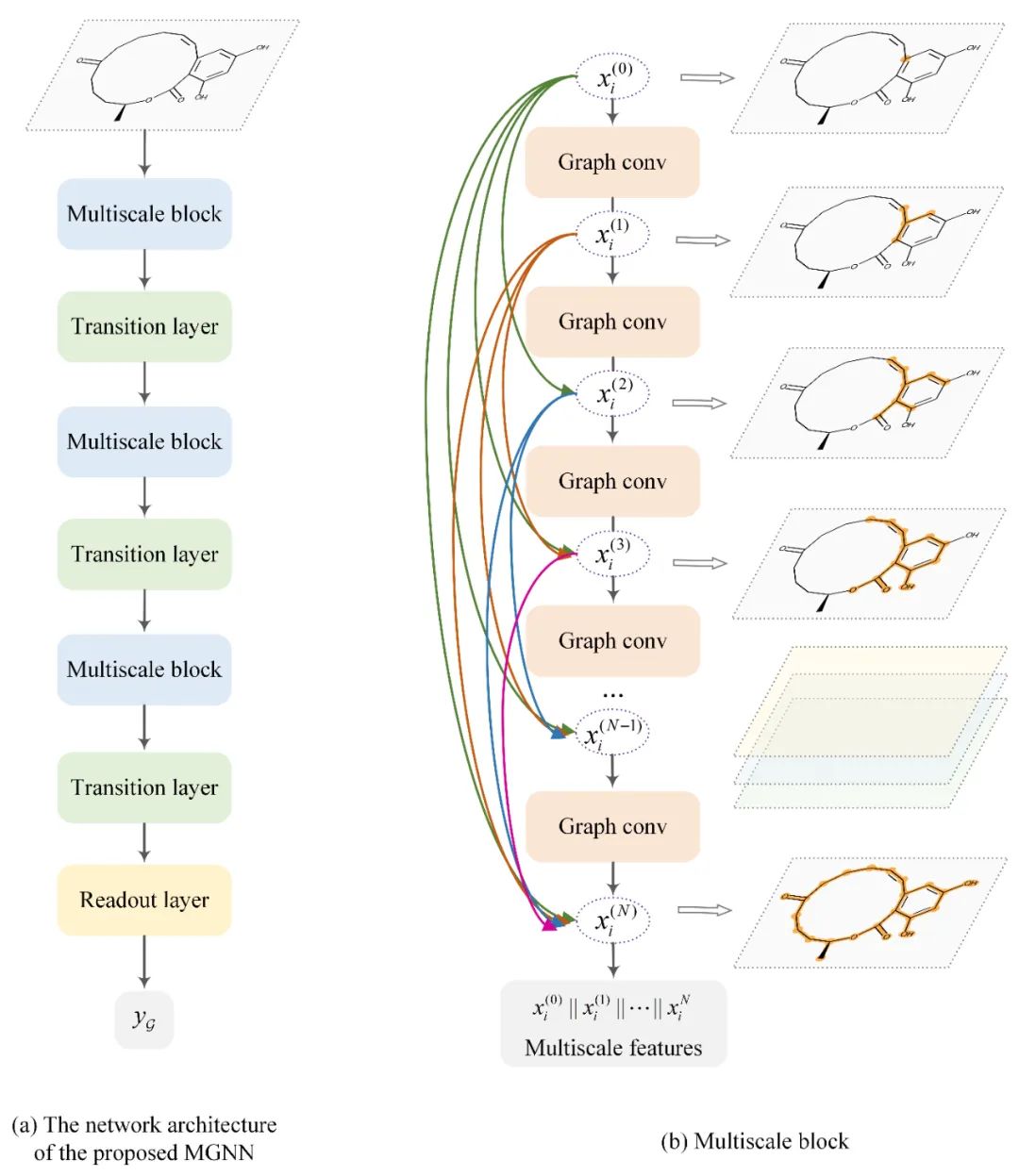
图3. MGNN的设计原理
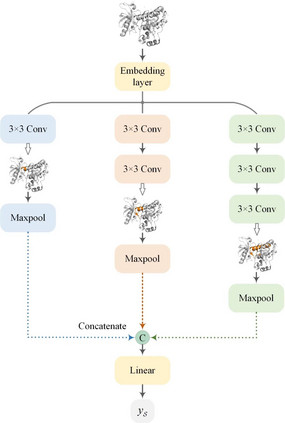
基于多尺度卷积神经网络(MCNN)的靶标特征提取 MCNN使用三个不同深度的CNN来提取不同尺度的靶标特征(图4)。在该设计中采用了三个较浅的CNN,这是因为在药物-靶标相互作用发生在蛋白质口袋,所以靶标的局部特征在DTA预测中比全局特征要更加重要,而设计深层CNN使其感受野覆盖整个蛋白质可能会引入噪声,从而降低DTA的预测精度。
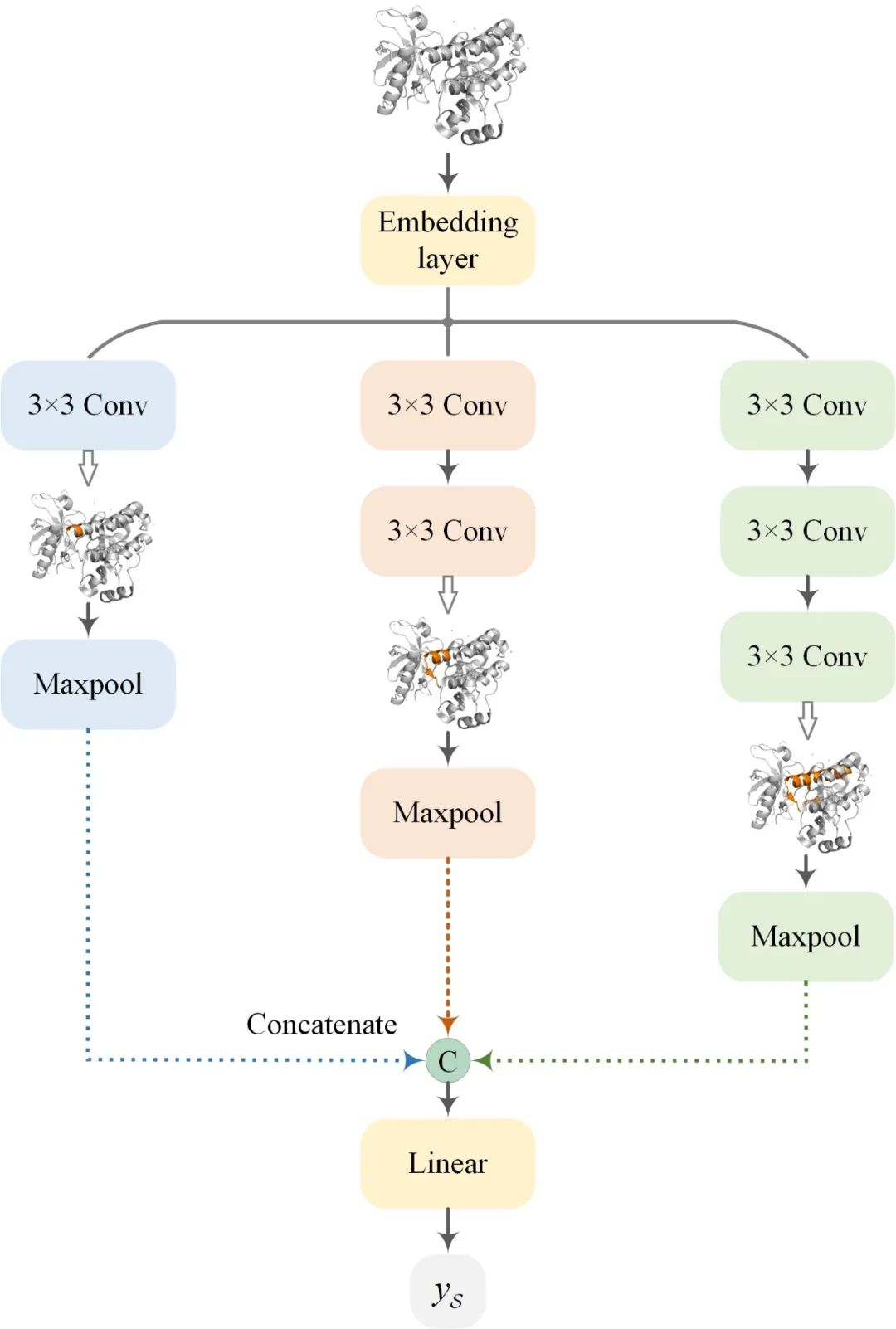
图4. MCNN的设计原理
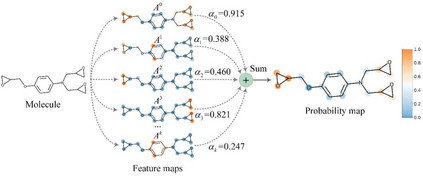
模型可视化方法 作者开发了一种基于亲和力激活的可视化方法Grad-AAM(图5)。Grad-AAM利用预测的亲和力在最后一层图卷积所产生的梯度大小来计算不同原子在DTA预测中的贡献。通过Grad-AAM,我们可以了GNNs是如何做出决策的。
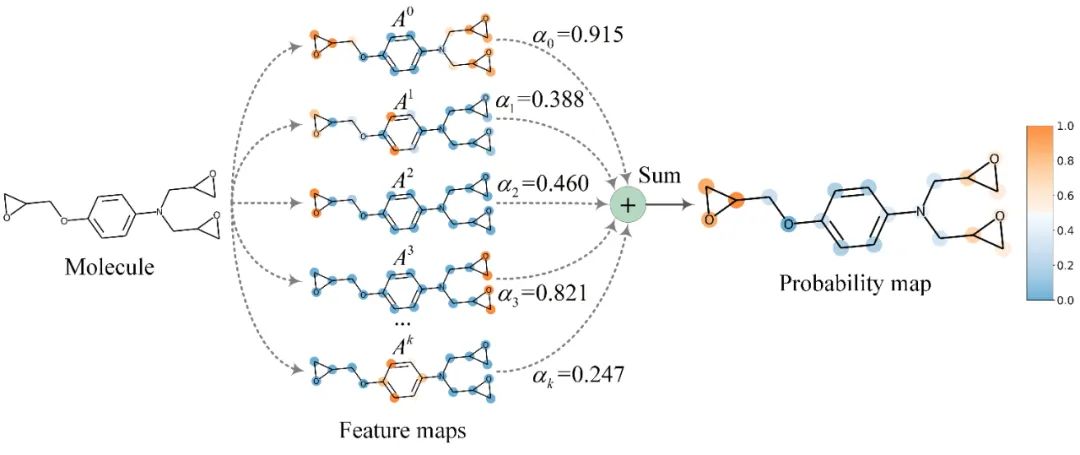
图5. Grad-AAM用于模型可视化
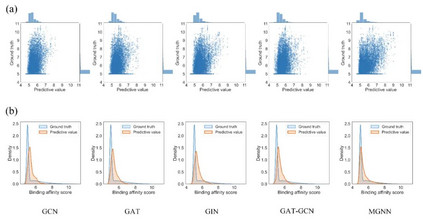
MGraphDTA提高DTA预测模型的泛化能力 我们在7个开源DTA数据集上对MGraphDTA进行测试,MGrahpDTA在所有指标上均优于已有方法,证明该论文所提策略的有效性。同时,当测试集中的药物不存在于训练集时,MGraphDTA仍然表现出良好的泛化能力。使用MGNN来提取药物的特征还有利于减小模型偏差,使模型预测的DTA分布更接近于真实分布(图6)。
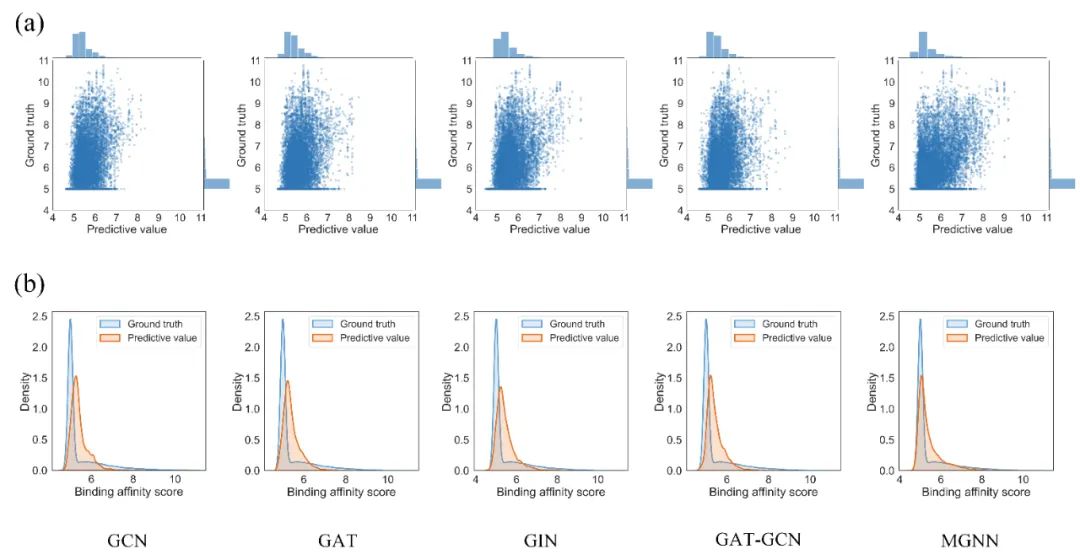
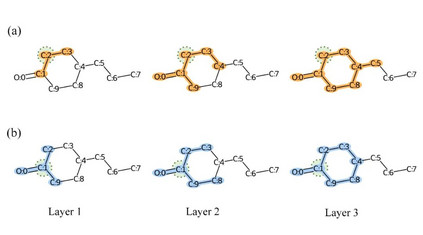
图6. 不同GNNs模型在Davis数据集上的预测分布和真实分布 MGraphDTA缓解图神经网络过平滑问题 当GNNs的网络深度过深时,会出现过平滑问题。具体而言,随着层数的增加,GNNs的感受野越来越大,这就导致了以不同原子为中心提取的化合物亚结构差异越来越小。如图7所示,但GNN只有一层时,以C1和C2原子为中心提取的化合物亚结构差异较大,但是三层GNN提取得到的化合物亚结构差异已经很小了。由于MGNN提取了化合物的多尺度特征,因此较小的化合物亚结构也可以得到保留,从而解决了过平滑问题。
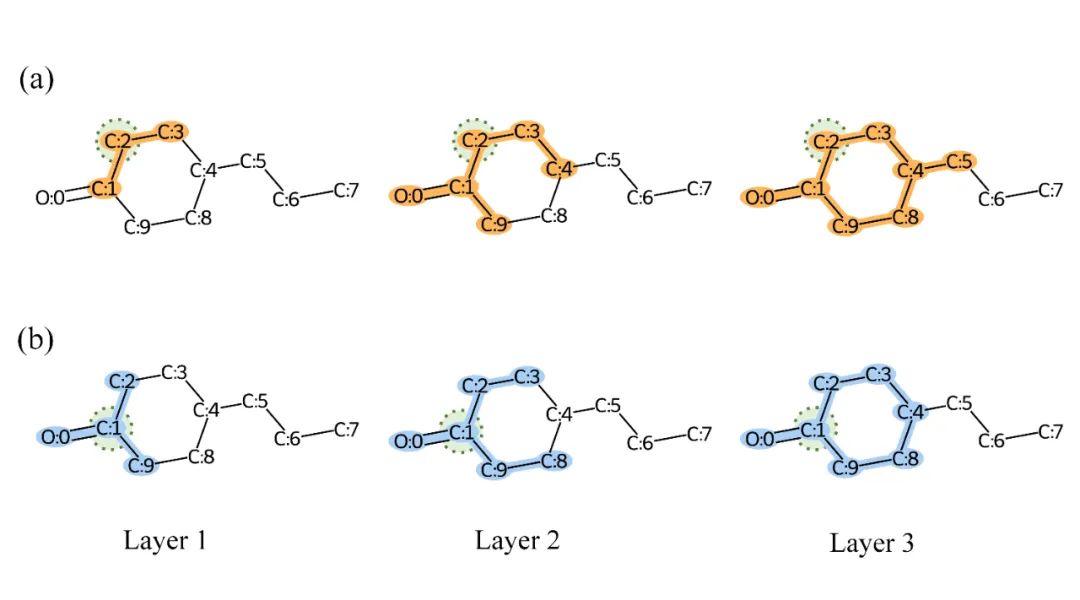
图7. GNNs过平滑问题
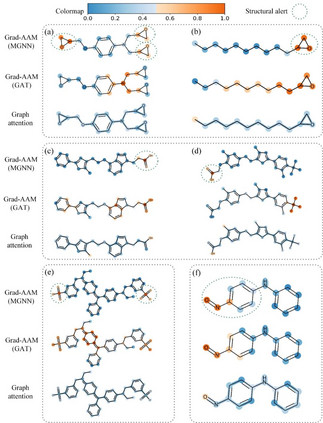
6、基于Grad-AAM的可视化结果 我们利用Grad-AAM在毒性数据集ToxCast上进行可视化测试,结果表明MGraphDTA能够捕捉到与毒性相关的官能团(图8),其可视化效果优于基于注意力机制的可视化效果。这些可视化结果表明Grad-AAM可能作为一种强有力的可视化工具帮助化学家分析药物-靶标相互作用的化学机理。
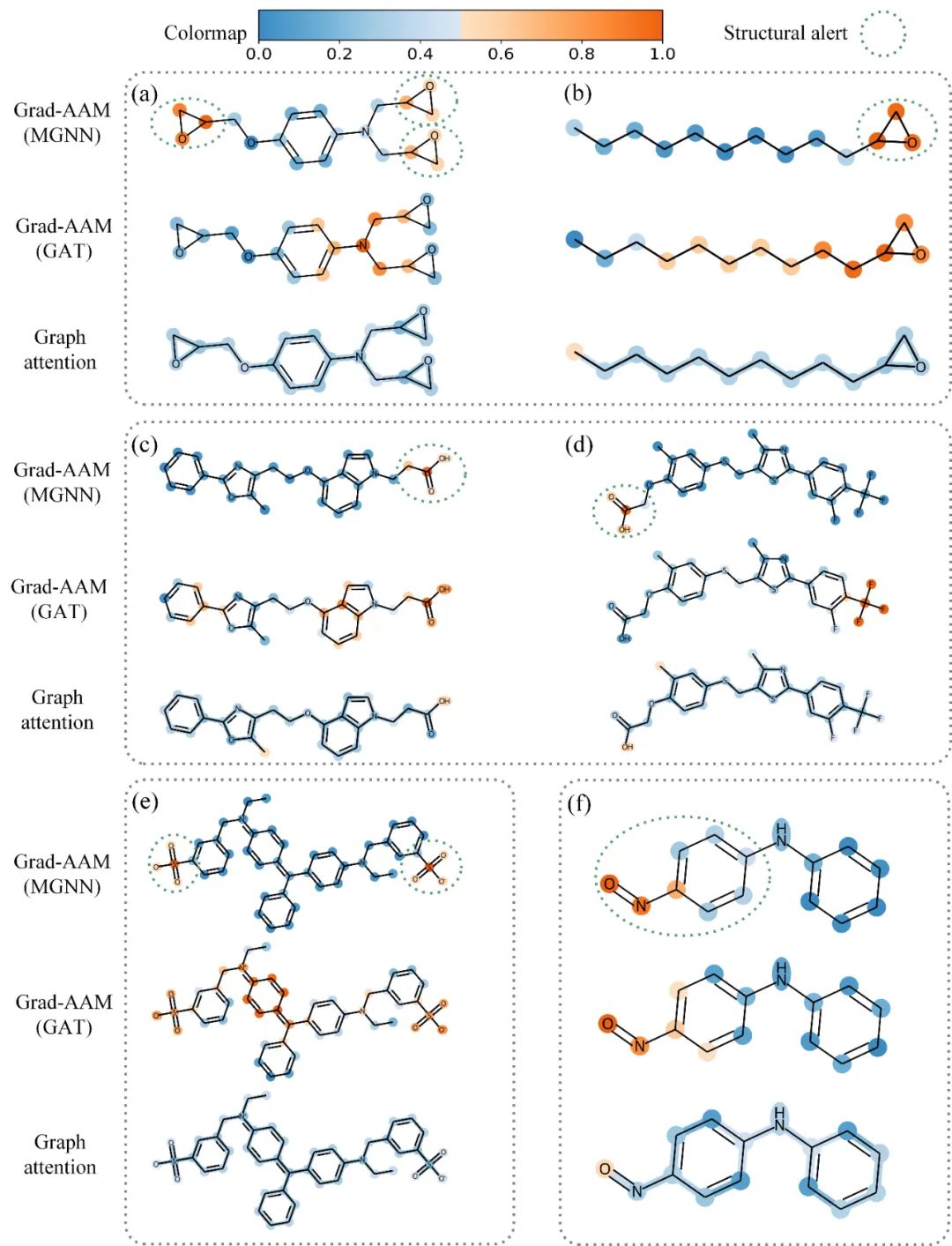
图8. 基于Grad-AAM的可视化结果 4 结论与展望 本文基于化学直觉设计一种新颖的图神经网络框架MGraphDTA。MGraphDTA通过引入稠密连接将图神经网络拓展了27层的深度,使其能够通过捕捉化合物的全局结构和局部结构,从而提高了模型的泛化性能。同时,作者设计了一种基于亲和力激活的可视化方法Grad-AAM,可视化结果表明Grad-AAM可能作为一种强有力的可视化工具帮助化学家分析药物-靶标相互作用的化学机理
致谢
该研究得到了国家自然科学基金面上项目(No. 62176272)等项目的支持。
Authors: Ziduo Yang†, Weihe Zhong†, Lu Zhao, Calvin Yu-Chian Chen* Title: MGraphDTA: Deep Multiscale Graph Neural Network for Explainable Drug-target Binding Affinity Prediction Published in: Chemical Science, doi: 10.1039/d1sc05180f