

在过去几十年里,计算机在化合物逆合成领域的应用取得了巨大的发展。本文分享了当前已有的计算机辅助逆合成工具,并讨论了目前逆合成预测方法所面临的挑战和机遇[1-2]。
背景
逆合成的概念最初由Corey在20世纪60年代提出,用来描述通过断键将一个复杂的目标分子还原为一个简单前体的迭代过程,即从产物出发, 搜索可能的前体,最终找到可商购获得的反应物。最初的逆合成预测主要依赖于经验丰富的化学家的已有知识或经验,缺乏系统的方法,所以难以满足一直不断快速增长的新有机化学反应的需求,也无法用于预测新的化学反应。近年来,得益于快速发展的数据驱动模型以及大型反应数据库,化学家和计算机科学家在计算机辅助逆合成方面取得了显著的进步。 本文是对目前人工智能辅助逆合成方法的综述,介绍并评估了在过去五年中开发的计算机辅助逆合成工具,结尾讨论了逆合成研究的未来发展方向以及目前面临的挑战。
逆合成基本理论
计算机辅助化合物合成路线规划(CASP)的框架通常由四部分组成,包括:建议断开的模板库、根据目标分子生成候选反应物的递归模板应用模块、具有市售起始材料的化学物数据库、以及单步或多步合成步骤评分方法。在过去十年中,由于大型反应数据库的建立和数据驱动计算工具的进步,逆合成预测方法取得了重大进展[3],本节主要介绍了逆合成方法中的一些关键概念。
反应的模板
反应模板是用于确定反应物如何通过断键转化为产物的一组规则。以前的反应模板是化学家定义和手动编码的。例如Chematica,它是目前商业上可用的、手动编码的反应库之一,其涵盖了大多数已知的反应规则,但手动编码现在所有的化学反应是一项艰巨的任务。一种更先进的反应编码方法是通过原子-原子映射算法提取反应中心,从而识别反应物和产物原子之间的对应关系。对于给定的反应,将一组键连接性改变的原子确定为反应中心,然后通过算法提取反应中心和相邻原子并进行泛化,最终形成相应的逆合成模板(图1)。
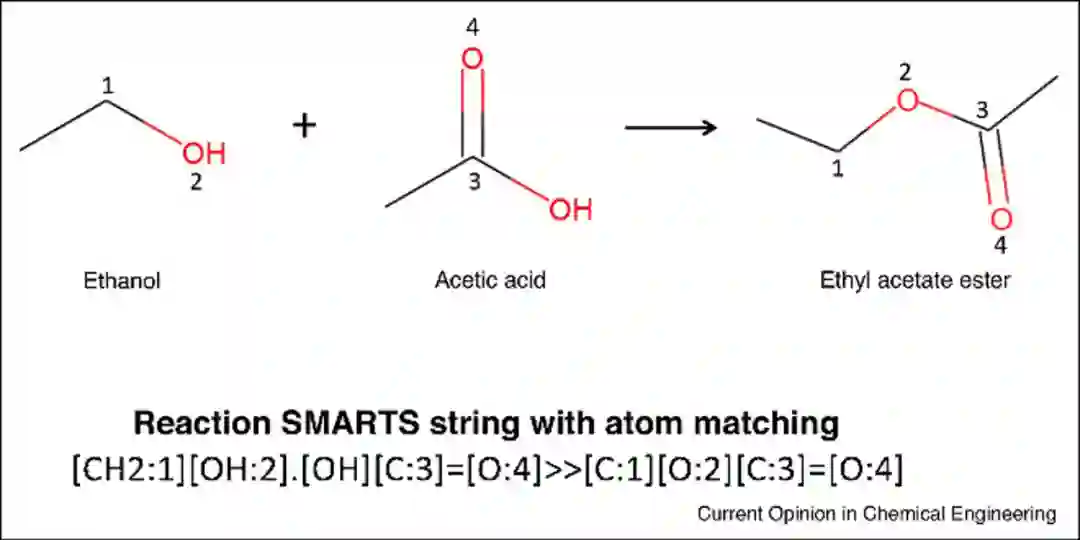
图1** 酯化反应SMARTS模板**[2]****逆合成方法评估
评估单步逆合成模型性能的一个常见指标是 Top-N 准确度,即在前n条推荐结果中出现数据集中记录的标准前体数量的百分比。该指标需要分子结构的精确匹配。此外,也可以通过分子相似性评分进行评估,即相似性得分为 1 表示结构相同。但近期研究人员们认为这种指标用来评估模型性能并不适合。 基于模板的逆合成模型
基于模板的逆合成模型是通过解决子图同构问题将目标分子与整个模板库匹配以获得候选反应物(图2)。该方法通常需要对反应数据库进行详尽的枚举,因此需要辅以高效的图论算法和虚拟筛选技术。
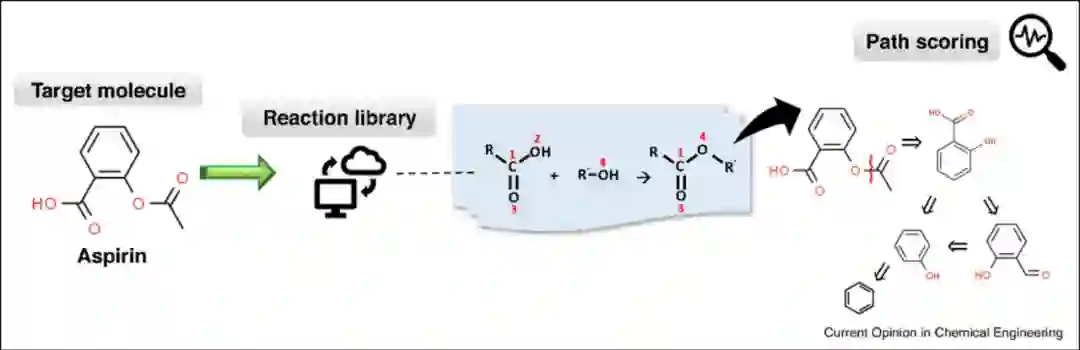
图2** 基于模板的逆合成模型(阿司匹林逆合成预测)[2]**
同时,为了量化分子结构的可合成性,在模型中引入可合成性的评价指标。传统的评价指标依赖于生成的SMILES字符串的长度,旨在将目标分子分成尽可能小的反应物。最近新的评价指标有:合成可行性分数(SASCORE)、SCScore、DRSVM以及分子复杂度等[4]。 基于模板的方法准确性较高,这主要是因为该方法的可解释性,以及该方法需要提供指定的化学前体。然而,该方法对计算能力需求很高,并且在模板库之外的泛化能力有限。
机器学习在基于模板的逆合成模型中的应用
基于模板的逆合成模型的研究主要集中在克服因枚举反应模板而导致的高计算成本。为了解决这个问题,研究人员利用机器学习只选择相关的模板,而不是使用完整的模板库。这类基于模板的模型被称为“聚焦模板应用程序”。该方法可以减少原来基于模板的方法的高计算强度,同时保持生成结果的化学可解释性,但是仍然无法预测反应模板库外的新反应。
无模板的逆合成模型
最近,无模板的逆合成方法引起了越来越多的关注,因其避免了高计算成本的子图匹配问题。该方法利用分子的文本表示(SMILES或InChI)将逆合成问题转换为序列到序列(seq2seq)预测问题,即将产物的SMILES字符串转化为反应物的SMILES字符串。 无模板的逆合成模型显示出优于基于模板方法的两个优势。首先,该模型可以隐式地学习反应规则和候选排名指标,从而避免了反应复杂度排名指标的使用。其次,无模板模型更加简便,手动编码反应模板仍然是基于模板的方法的主要缺点之一。 但无模板方法在逆合成预测中仍然相对较新。该领域的最新发展依赖于仅包含注意力机制的NLP模型,例如Transformer。并且目前无模板方法的预测准确率仍稍落后于基于模板的方法。解决无模板方法中无效 SMILES 字符串的输出是提高该方法准确性的关键。
小结
机器学习通过从丰富的化学知识中学习,为多步逆合成规划框架做出了贡献。然而,基于机器学习的逆合成模型的性能在很大程度上取决于反应数据库的质量。从文献中收集的反应数据可能具有噪声大且不准确的问题,所以高质量的数据库将加速逆合成模型的进一步发展。 同时单纯基于数据驱动的逆合成模型有时会缺乏可解释性。尤其是基于序列的无模板方法可能会忽略断键背后的重要化学意义,这会导致解码生成不可行的逆合成路径。因此提高机器学习可解释性的方法可能是应对这一挑战的解决方案。同时,由于大多数反应数据以常见反应类型为主,罕见反应在数据集中代表性不足,因此机器学习模型会从数据集中出现次数多的断键规则中学习,从而忽略其他罕见但更加简单的反应途径的断键可能性。减少模型偏差的一个解决方案是将数据驱动方法与基于模板的方法相结合。这两种方法的结合可以提高模型的可解释性,并为逆合成问题提供新颖有效的解决方案。 任何计算机辅助化合物逆合成路线规划,都应通过实验验证,以确定计算机辅助逆合成方法的真实性能。此外,训练数据中缺乏实验条件是当前逆合成方法的另一挑战。 人工智能驱动的药物合成给社会带来极大的便利。在数字化趋势的推动下,人工智能有望成为建立自动化化学合成系统的基本组成部分,最终成为未来的“机器人化学家”。
**参考文献 **
[1] Ucak U , Ashyrmamatov I , Ko J , et al. Retrosynthetic reaction pathway prediction through neural machine translation of atomic environments. Nature Communications, 2022, 13(1). DOI: 10.1038/s41467-022-28857-w. [2] Sun Y, Sahinidis N V. Computer-aided retrosynthetic design: fundamentals, tools, and outlook. Current Opinion in Chemical Engineering, 2022, 35: 100721. [3] Segler M , Preuss M , Waller M P . Planning chemical syntheses with deep neural networks and symbolic AI. Nature, 2018, 555(7698):604-612. [4] Thakkar A , V Chadimová, Bjerrum E J , et al. Retrosynthetic accessibility score (RAscore) – rapid machine learned synthesizability classification from AI driven retrosynthetic planning. Chemical Science, 2021. DOI: 10.1039/D0SC05401A.
供稿:张红文
校稿:张梦婷/谢金欣编辑:王思雨华东理工大学/上海市新药设计重点实验室/李洪林教授课题组▼招聘博后▼华东理工大学李洪林教授团队诚聘博士后



