论文剖析
生物信息学|逆合成预测的学习图模型 ///////////////
- 摘要
逆合成预测是有机合成中的一个基本问题,其任务是识别可用于合成目标分子的前体分子。为这一任务建立神经模型的一个关键考虑因素是将模型设计与化学家采用的策略相一致。基于这一观点,本文介绍了一种基于图的方法,该方法利用了前驱体分子的图拓扑结构在化学反应中基本不变的观点。该模型首先预测了一组将目标转化为不完整分子的合成子。接下来,该模型通过附加相关的离去基来学习将合成子扩展成完整的分子。这种分解简化了体系结构,使其预测更具可解释性,也可以手动修正。我们的模型达到了53.7%的top1精度,优于以前的无模板和基于半模板的方法。 2. 介绍
逆合成预测,首先由E.J.Corey提出是有机合成中的一个基本问题,它试图识别合成目标分子的一系列化学转化。在单步逆合成中,任务是识别一组给定一个目标的反应物分子。除了简单的反应之外,许多涉及复杂有机分子的实际任务,即使对专家化学家来说也是困难的。因此,需要进行大量的实验探索来弥补分析方法的不足。这激发了人们对计算机辅助逆合成的兴趣。
在计算上,主要的挑战是如何探索能够产生目标分子的反应的组合空间。在很大程度上,以前的逆合成预测方法可以分为基于模板的和无模板的方法。基于模板的方法将目标分子与一组大型模板进行匹配,这些模板是在化学反应过程中突出变化的分子子图模式。尽管这些方法具有可解释性,但它们并不能推广到新的反应中。无模板的方法通过学习从SMILES的产物表示到反应物的直接映射来绕过模板。尽管这些方法具有更大的泛化潜力,但它们会逐个生成反应物SMILES特征,从而增加了生成的复杂性。
在建立逆合成模型时的另一个重要考虑因素是将模型设计与专家化学家采用的策略保持一致。这些策略受到化学反应的基本性质的影响,独立于复杂性水平:(i.)产物原子总是反应物原子的子集,和(ii)从产物到反应物,分子图拓扑在很大程度上没有改变。例如,在标准的逆合成数据集中,产物中只有6.3%的原子的连通性发生了变化。
这种考虑在最近的基于半模板的方法中得到了更多的关注,该方法分两个阶段生成反应物:(i.)首先识别被称为合成子的中间分子,(ii)然后通过连续生成原子或SMILES特征将合成子完成成反应物。我们的模型graphretro也使用了类似的工作流。然而,我们通过从预先计算的词汇表中选择被称为leaving groups的子图来避免完成合成子的顺序生成。这个词汇表是在预处理过程中通过提取合成物和相应反应物之间不同的子图来构建的。该词汇表的大小较小(USPTO-50k为170),表明存在显著的冗余,同时覆盖了99.7%的测试集。在这些子图的水平上进行操作,大大降低了反应物生成的复杂性,并提高了经验性能。这个公式还简化了我们的体系结构,并使我们的预测更加透明、可解释和易于手动校正。
评估合成模型的基准数据集是USPTO-50k,它包含了10个反应类别的50000个反应。数据集包含了一个预测编辑(化学反应前后发生变化键和原子)的意外快捷方式,因为在75%的情况下,具有原子映射1的产品原子是编辑的一部分,允许依赖于原子位置的预测高估性能。我们规范化产品SMILES并重新映射现有数据集,从而删除快捷方式。在这个重新映射的数据集上,当反应类别未知时,graphretro方法达到了53.7%的top-1精度,优于无模板和基于半模板的方法。 3. 相关工作
现有的逆合成预测机器学习方法可以分为基于模板的方法、无模板的方法和最近的基于半模板的方法。
基于模板:模板要么由专家手工制作,要么从大型数据库中算法提取。由于涉及子图匹配过程,应用大型模板集的代价很高。因此,基于模板的方法利用不同的方式对模板进行优先排序,通过学习模板集上的条件分布,根据先前反应的分子相似性对模板进行排名,或使用逻辑变量直接对模板和反应物的联合分布建模。尽管具有可解释性,但这些方法无法在其规则集之外泛化。
无模板:无模板方法利用神经机器翻译结构学习从产物到反应物的直接转化。将分子线性化并不利用固有的丰富的化学结构。此外,反应物的SMILES是从头开始产生的。人们试图通过添加语法校正器和混合模型来提高建议的多样性,但在标准逆合成数据集上的性能仍然低于基于模板的方法。Sun等人利用基于能量的模型制定了逆合成,并附加了参数化和损失项,以加强正向(反应预测)和反向(逆合成)预测之间的对称性。
基于半模板:我们的工作与最近提出的基于半模板的方法,首先识别合成子,然后扩展合成子到反应物。为了降低反应物生成的复杂性,我们使用从预先计算的词汇表中选择的称为leaving groups的子图来完成合成子。这允许我们将合成子扩展视为一个分类问题,而不是一个生成问题。我们还利用了可能的编辑之间的依赖性图,并使用该图上的消息传递网络(MPN)更新编辑预测。与以前的基于半模板的方法相比,这两种创新的性能分别提高了4.8%和3.3%。
反应中心识别覆盖了少量参与反应的原子。我们的工作还与预测反应结果的模型有关,通过学习对位于反应中心的可能性进行排序。识别反应中心的任务与在我们的公式中推导合成子的步骤有关。我们还利用可能的编辑之间的依赖图,并使用此图上使用MPN更新编辑预测。 4. 模型设计
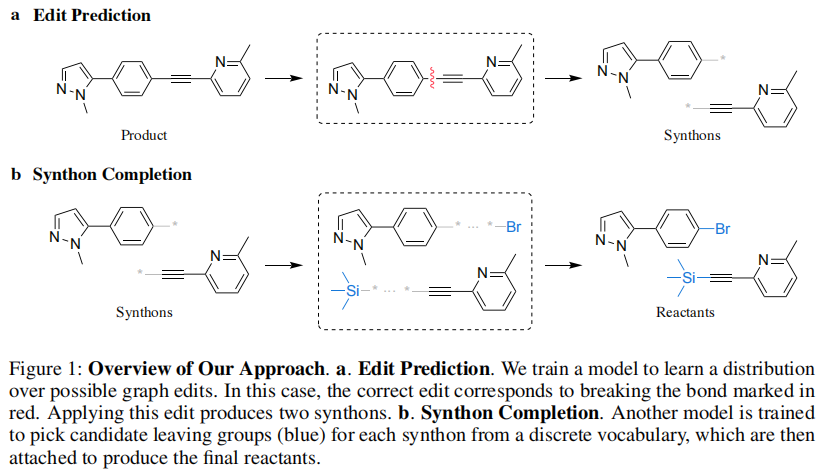
我们的方法利用了图拓扑从产物到反应物基本不变的特性。为了实现这一点,我们首先从被称为合成子的产物中获得合适的构建块,然后通过添加leaving groups,将它们完成成有效的反应物。我们首先训练一个神经网络来预测可能的编辑的分数。然后将得分最高的编辑应用于产物,以获得合成子。由于唯一的leaving groups的数量较少,我们将leaving groups的选择建模为预先计算的词汇表上的一个分类问题。为了产生候选反应物,我们通过化学约束规则将预测的离去基附加到相应的合成物上。图1概述了整个过程。
- 结论
以往的单步逆合成方法要么限制对模板集的预测,要么对分子图结构不敏感,要么从头开始生成分子。我们通过引入一个受化学家工作流程启发的基于图的半模板模型来解决这些缺点,增强了逆合成模型的可解释性。给定一个目标分子,我们首先确定合成的构建模块,然后实现为有效的反应物,从而避免从零开始生成分子。我们的模型在基准数据集上比以前的半模板方法有显著的优势。未来的工作目标是扩展该模型,使其从多个合成物中实现单一反应物,并引入更多具有化学意义的成分,以提高此类逆合成预测工具与从业人员专业知识之间的协同作用。 原论文名称: Learning Graph Models for Retrosynthesis Prediction