分子表示学习(Molecular representation learning, MRL)是建立机器学习与化学科学联系的关键步骤。特别是,它将分子编码为保留分子结构和特征的数值向量,在此基础上可以执行下游任务(如性能预测)。近年来,MRL取得了相当大的进展,特别是在基于深度分子图学习的方法中。在这项综述中,我们系统地回顾了这些基于图的分子表示技术。具体来说,我们首先介绍了二维和三维图形分子数据集的数据和特征。然后,我们总结了专门为MRL设计的方法,并将其分为四种策略。此外,我们还讨论了MRL支持的一些典型的化学应用。为了促进这一快速发展领域的研究,我们也在论文中列出了基准和常用的数据集。最后,我们分享了对未来研究方向的思考。
机器学习和化学科学之间的相互作用受到了这两个领域研究人员的极大关注。它在包括分子性质预测在内的各种化学应用中取得了显著的进展[Guo et al., 2020; Sun et al., 2021; Yang et al., 2021b; Liu et al., 2022b],反应预测[Jin等人,2017;Do等人,2019],分子图生成[Jin et al., 2018a; Jin et al., 2020b]以及药物-药物相互作用预测[Lin等人,2020]。分子表示学习(MRL)是弥补这两个领域差距的重要步骤。MRL的目标是利用深度学习模型将输入的分子编码为数值向量,保存有关分子的有用信息,并作为下游(机器学习)应用的特征向量。早期的分子表示学习方法使用一般表示学习模型来表示分子,而不需要明确地涉及领域知识。近年来,针对MRL专门设计了许多算法,这些算法可以更好地融合化学领域知识。在本文中,我们系统地回顾了这一快速发展的主题的进展,绘制了从结合分子结构的表示学习方法到同时结合领域知识的方法的路径。
动机1: 为什么分子表示学习很重要?
分子表示学习具有广泛的应用范围,与人们的生活密切相关。例如,通过wet-lab实验发现药物是非常耗时和昂贵的。随着深度学习的发展,大量的实验可以用机器学习模型来模拟。性质预测可以帮助识别具有目标性质的分子。反应预测可以预测主要产物。这大大减少了失败实验的数量。对于所有这些化学应用,MRL是深度学习模型成功的关键决定因素。
动机2: 为什么要用深度图学习来进行分子表示学习?
分子图自然地描述了具有丰富结构和空间信息的分子。分子本质上是原子和连接原子的键,这自然会导致它们自己的图表示。相对于基于分子的线状表示(即串),分子图为MRL模型提供了更丰富的信息。因此,基于图的MRL模型比基于序列的MRL模型发展得更快。此外,越来越多的通用图学习论文[Gilmer et al., 2017; Hu* et al., 2020; You et al., 2020]也使用分子图数据集来检查他们算法的性能。
这项工作的主要贡献总结如下:
- 我们系统回顾了基于各种分子输入的基于图的MRL模型的最新进展,并总结了专门针对MRL设计的策略。
- 为了鼓励对该主题的可重复性研究,我们总结了各种下游应用中的代表性基准和常用数据集。
- 我们讨论了二维和三维分子图的局限性,并分享了我们对未来MRL研究方向的想法,以供社区参考
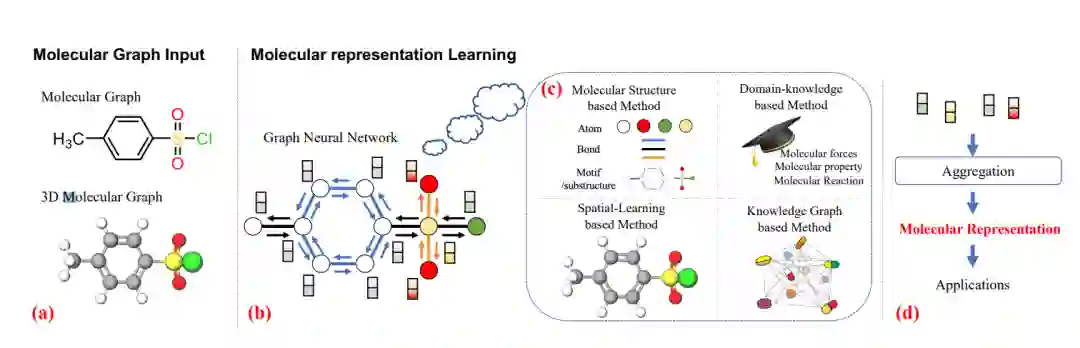
基于图的分子表示学习综述: (a) 两个分子图; (b) 图神经网络的一般学习过程; (c) 提出了四种基于图的分子表示学习方法; (d) 聚合原子表示以获得分子表示的过程。
数据表示
传统上,研究人员使用固定的指纹特征提取规则来识别每个分子的重要信息,并将这些手工制作的信息输入线性分类/回归头部进行下游任务。这需要大量的时间来确定和计算最相关的功能,而设计的功能仍然不能支持所有的任务。为了避免这些问题,大多数深度学习模型都是用来自动学习分子特征的。两种分子表示被用作输入:分子图和分子序列。据此,开发了基于图和基于序列的模型,以从不同的输入分子表示中学习。序列表示,如简化的分子输入行输入系统(SMILES) [Weininger et al., 1989]和自引用嵌入字符串(selfie) [Krenn et al., 2020]可以转换为分子图,但这种转换涉及大量领域知识。当我们以序列表示作为输入时,基于序列的学习模型不容易意识到这一知识。相反,图表示可以自然地在节点和边中包含额外的信息,这很容易被丰富的基于图的模型套件(例如,图神经网络)所利用。因此,在这次调研中,我们将重点放在图表示上,因为它现在使用的比较普遍。在本节中,我们将阐明分子图(不含空间信息)和三维分子图表示,如图1 (a)所示。对于每一种表示,我们都将分析其特点,并讨论其在深度学习模型中使用的用途和局限性。
3 方法
在本节中,我们从MRL的一般图神经网络开始。然后,我们讨论了专门为这项任务设计的方法,并将这些方法分为四种策略。这些特定的方法结合了化学相关信息,以不同的方式加强分子表征,从而导致更好的性能。表2列出了具有代表性的方法。
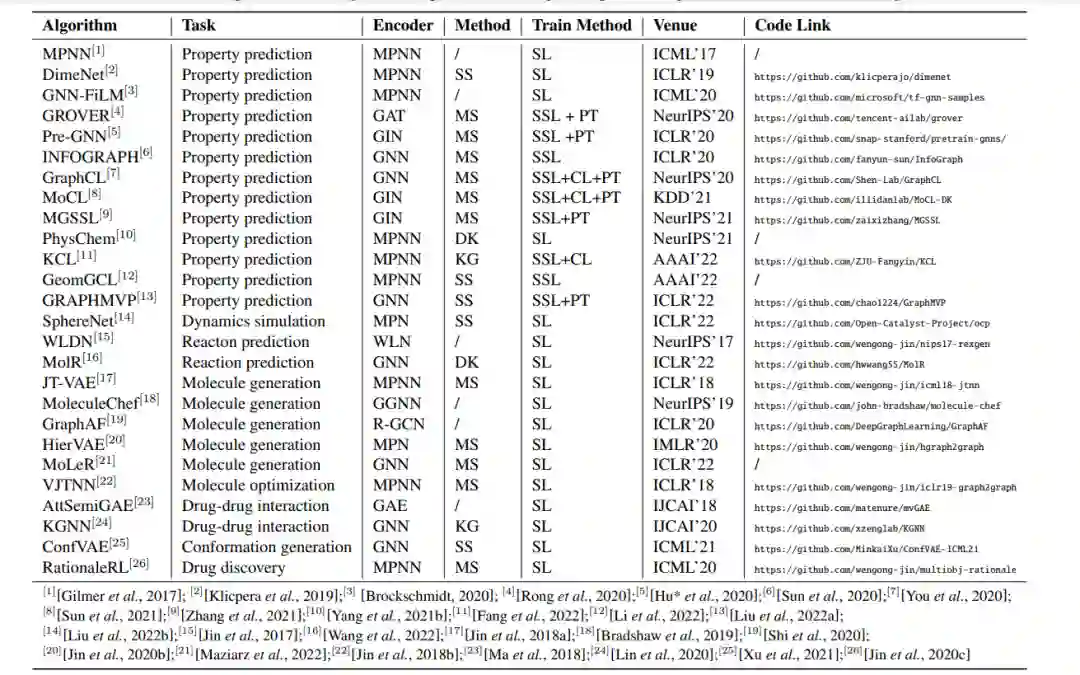
表一 一个具有代表性的基于图的分子表示学习算法列表。四种方法(MS, DK, SS, KG)分别对应第3节的四个部分。在这里,MS专门代表了分子子结构相关的方法。该表包括四种训练方法:自监督学习(SSL)、监督学习(SL)、预训练学习(PT)和对比学习(CL)。
3.1 基于分子结构的方法
基于图的MRL通常认为分子图与其他平面图是一样的。它只关注分子图的拓扑结构,而不关心分子图中包含的特殊子结构或性质。最近的研究已经看到了对自监督学习策略的尝试[Jin et al.,2020a],这促使模型更加关注图结构。PreGNN [Hu et al.,2020]利用上下文预测和节点/边缘属性掩蔽两种自监督策略对GNN进行预训练。与这种一般的无监督设计不同,GROVER [Rong et al., 2020]提出了分子特异性的自监督前训练方法:上下文属性预测和图形级motif预测。MGSSL[Zhang et al.,2021]还设计了一种基于主题的图自监督策略,在主题树生成过程中预测主题的拓扑和标签。INFOGRAPH[Sun et al., 2020]通过最大化整个图的表示和不同粒度的子结构之间的互信息来训练模型。
对比学习是一种常见的自监督学习策略,它利用数据增强使模型产生具有更好的泛化性、可迁移性和鲁棒性的图表示。GraphCL [You et al.,2020]提出了三种通用的图增强方法,也可应用于分子数据集。MoCL [Sun et al.,2021]提出了两种分子图增强方法:一种是用类似的物理或化学性质相关的子结构取代有效的子结构。另一种是改变一些普通的碳原子。分子2D和3D图形表示自然是分子的两种增强视图。利用这一特性,GeomGCL[Li et al.,2022]和GRAPHMVP[Liu et al.,2022a]通过对比学习训练模型。分子结构知识不仅用于自监督学习。基于Motif、substructure和scaffold的分子表征学习在分子生成中的应用[Jin et al., 2020c; Maziarz et al., 2022; Wu et al., 2022]也取得了具有很好的性能。
领域知识的方法
将深度学习与分子科学相结合是分子表示学习的重要组成部分。在模型设计中引入化学领域知识是提高性能的有效途径。Yang等人[Yang等人,2021b]提出了一个新的模型,PhysChem,它由物理学家网络(PhysNet)和化学家网络(ChemNet)组成。PhysNet通过神经网络学习分子构象,ChemNet通过神经网络学习化学性质。通过融合物理和化学信息,PhysChem获得了性能预测任务所需的性能。PAR[Wang et al., 2021]涉及任务信息,提出了一种属性感知的嵌入方法。Wang等人[Wang等人,2022]的灵感来自化学反应中反应物和生成物之间的等价关系。他们提出,MolR,在嵌入空间中保持等价关系,这意味着使反应物嵌入和乘积嵌入的和相等。MolR在各种下游任务中实现SOTA性能。
基于空间学习的方法
空间信息尤其是几何信息受到广泛关注,越来越多地涉及到分子表示学习过程中,特别是当模型需要学习原子上的力或能量时。DimeNet [Klicpera et al., 2019],GemNet [Klicpera et al., 2021a]和定向MPNN [Klicpera et al., 2021b]提出了定向消息嵌入。虽然他们仍然以二维分子图作为输入,但他们不仅考虑了原子之间的距离,还考虑了空间方向,这是由原子的二维坐标计算的。它们利用方向信息,根据原子之间的角度转换信息。利用球面贝塞尔函数和球面谐波,可以有效地联合提出距离和角度。一般来说,二维图强调拓扑信息,而三维几何图更注重能量。GeomGCL [Li et al., 2022]计算确定的几何因子(角度和距离),并利用径向基函数获得几何嵌入。GRAPHMVP [Liu et al., 2022a]采用3D构象,通过3D GNN模型学习分子表示。为了完成三维图结构的识别,SphereNet [Liu et al., 2022b]设计了一个球形消息传递作为三维分子学习的强大方案。
基于知识图谱的方法
知识图谱是一种将分子结构不变但丰富的外部知识引入模型的有效策略。与以往的方法不同,KGNN [Lin et al., 2020]和MDNN [Lyu et al., 2021]探索以分子为节点,以分子之间的连接关系为边的知识图谱。这样,通过知识图谱结构而不是分子结构来学习分子表示。Fang等[Fang et al., 2022]构建了一个化学元素知识图,由(化学元素、关系、属性)形式的三元组构成,如(Gas, isStateOf, Cl)。他们建议使用这种KG来增加分子中的节点和边,并利用对比学习来最大化分子图的两种观点之间的一致性。
4 应用
在这里,我们介绍了几个代表性的应用和算法,以解释如何设计模型来处理基于MRL的特定应用。分子性质预测在药物发现中起着重要的作用,能够发现具有靶点性质的候选药物。通常,该任务包括两个阶段:生成固定长度分子表示的分子编码器和预测器。预测器根据学习到的分子表示预测分子是否具有目标性质或预测分子对目标性质的反应。性能预测结果可以直接反映学习到的分子表示质量。因此,性质预测问题得到了研究者的广泛关注。越来越多的通用图学习论文[Hu* et al., 2020; Gilmer et al., 2017; Brockschmidt, 2020; You et al., 2020]利用分子图数据集和属性预测任务来检查其算法的性能。首先提出了针对MRL的分子深度学习方法,并将其应用于本课题。MolR [Wang et al., 2022]提出了一种通过保持分子反应在嵌入空间中的等价关系来学习分子表征的新方法,该方法也首先应用于性质预测任务。此外,现有的分子数据集不足是化学领域普遍存在的问题。Guo等人[Guo et al., 2021]和Wang等人[Wang et al., 2021]提出了元学习方法来处理性质预测中的这个问题。
药物发现的关键挑战是找到具有靶标性质的靶标分子,这在很大程度上依赖于领域专家。分子生成是为了使这个过程自动化。完成这项任务需要两个步骤:一是设计一个编码器,以连续的方式表示分子,这有利于优化和预测性质;另一种是提出一种解码器,将优化后的空间映射到具有优化特性的分子图上。由于SMILES 不是用来捕获分子相似性的,分子生成模型大部分时间直接在分子图上操作。为了避免无效状态[Jin et al., 2018a],大多数工作都是通过子结构生成图的子结构,而不是通过节点生成图的子结构。JT-VAE [Jin et al., 2018a]和VJTNN [Jin et al., 2018b]基于图中的子结构,首先将分子图分解为连接树。然后他们用神经网络对树进行编码。接下来,他们重建连接树,并将树中的节点组装回原始的分子图。HierVAE [Jin et al., 2020b]基于基序分层生成分子图。MoLeR [Maziarz et al,2022]在生成过程中保持支架结构,并依靠基序生成分子。GraphAF [Shi et al,2020]利用流动模型生成分子图。MoleculeChef [Bradshaw et al,2019]是一种用于生成可合成分子的模型。它首先生成反应物分子,然后利用分子transformer (Schwaller et al., 2019)模型生成目标分子。
反应的预测
反应预测和反合成预测是有机化学的基本问题。反应预测是指利用反应物预测反应产物。反合成预测过程与反应预测相反。以SMILES为输入时,将反应预测任务作为翻译任务。以分子图为输入时,分为反应预测和反合成预测两步。与WLDN [Jin et al., 2017]和WLDN++ [Coley et al., 2019]一样,该模型需要先预测反应中心,然后预测潜在产物,这是主要产物。与之前的工作不同,MolR [Wang et al., 2022]将反应预测的任务定义为一个排序问题。测试集中的所有产品都放在候选池中。MolR根据从给定的反应物集学习到的嵌入对这些候选产物进行排名。
药物之间相互作用
检测药物-药物相互作用(DDI)是一项重要的任务,可以帮助临床医生做出有效的决策和安排合适的治疗方案。准确的DDI不仅可以帮助药物推荐,还可以有效地识别潜在的不良反应,这对患者和社会都至关重要。AttSemiGAE[Ma et al., 2018]提出通过测量多种药物特征的药物相似性来进行DDI。SafeDrug [Yang et al., 2021a]设计了全局和局部两个模块,以完全编码药物分子的连通性和功能,从而产生DDI。KGNN [Lin et al., 2020]和MDNN [Lyu et al., 2021]都构建了药物知识图谱,以提高DDI的准确性。
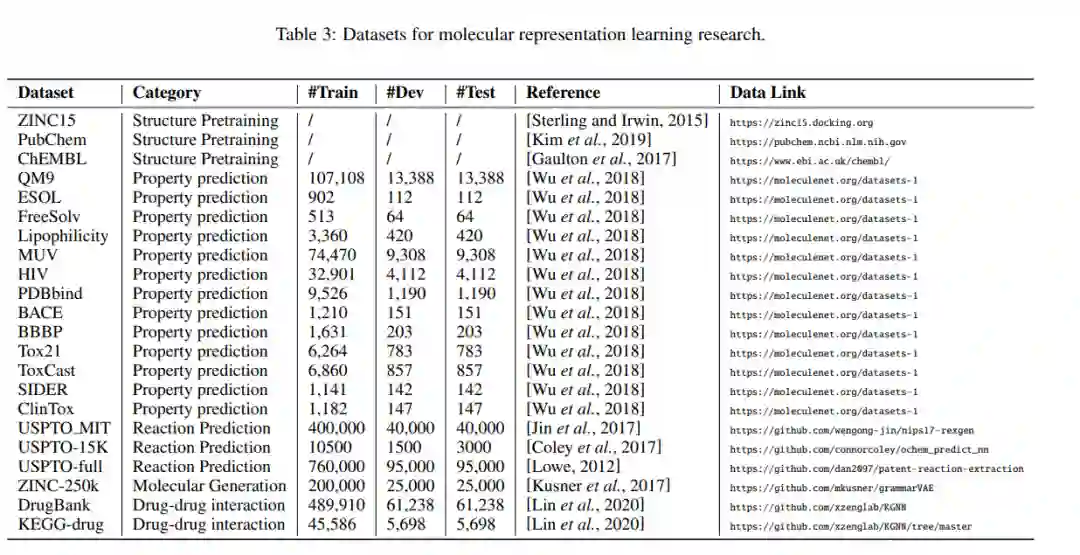
5. 数据集与基准
我们在表2中总结了代表性的分子表示学习算法。为方便查阅实证结果,每篇论文均附有代码链接(如有)。文中还列出了相应的任务、编码算法、方法和训练方法。这里,方法指定了我们在第3节中讨论的4种方法。对于训练方法,我们包括自我监督学习、监督学习、训练前学习和对比学习。除了算法,我们还在表3中总结了不同化学任务常用的数据集。