1. 背景
**
**
随着扩散模型杀入蛋白质设计领域,时隔几天,Grigoryan和Ingraham等人以Generate Biomedicines公司的名义发表了一个新的扩散蛋白生成模型Chroma。它的创新点为通过随机图神经网络的架构进行低复杂度的长距离推理,通过一个基于凸优化的结构模块从预测的残基间几何信息高效地预测蛋白质的三维结构,通过低温采样提升扩散效果,以及针对特定分布或性质的定向生成。 基于以上方法,Chroma可以使蛋白质设计成为外部约束下的贝叶斯推理过程,在生成大量可设计、新颖的蛋白基础上,可以根据根据对称性、motif、性质分类器等等进行蛋白工程,对于binder、抗体等设计应用。除此以外本文还进行了一些创新实验包括根据形状、自然语言指导生成,类似AIGC中的模型进行基于特定描述的图像生成一样,进行蛋白生成,这展示了Chroma的应用潜力。
2. 模型****方法
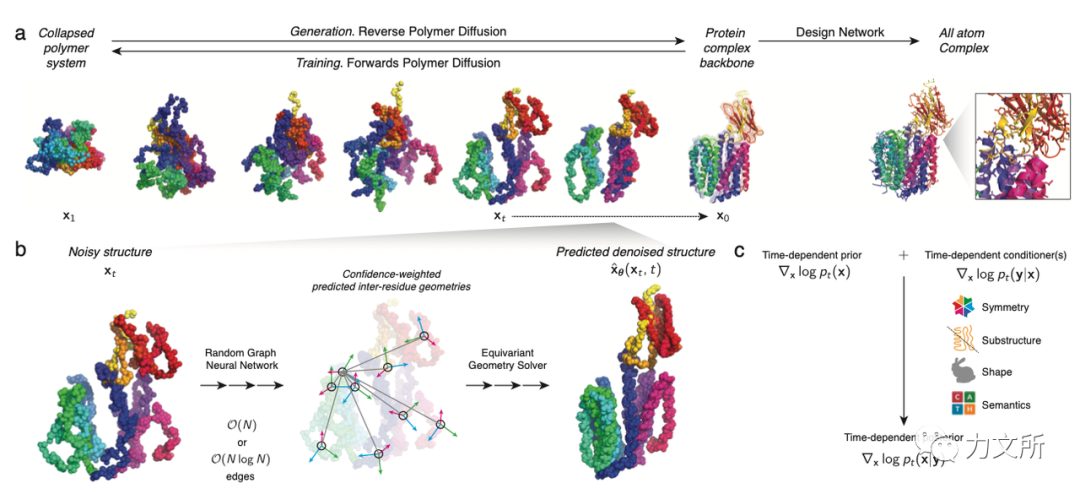
Chroma的主要架构如上图展示,其中多个模块都是在已有模型上的改进,比如随机图神经网络是在Ingraham2019年发表的GNN架构[1]上进行的改进,解码器借鉴了Terminator,侧链网络借鉴了Possu Huang 3DCNN的设置[2],坐标更新方法也仿照了Alphafold2的设置,但都进行了效率上的改进。 主要的扩散模型在a图展示,包括主链网络和设计网络两部分。蛋白结构在正向扩散过程中在被加以链和回旋半径约束的情况下加噪,并在逆向过程中去噪获得采样主链,并被最后输入到一个基于图的设计网络中采样获得蛋白序列和侧链构象。 b图展示了在扩散中使用的随机图神经网络和结构模块,最终从加噪结构中获得了去噪结构。 c图展示了与扩散模型先验结合的条件模型,其关键在于将给定约束条件的蛋白质条件分布的扩散过程加入来自外部分类器的梯度,本文这里的外部分类器包含范围广泛,指任何从加噪样本预测标签的监督模型,具体属性见下文。
2.1 结构约束扩散模型现有的一些蛋白扩散模型方法会在二面角、接触图上等低维度信息上进行,本文指出由于高斯噪音方法属于无关扩散,在三维信息上进行高斯噪音去噪需要模型从头学习这些基本的结构关系,而这往往又是不理想的。噪声损害了蛋白结构都会天然满足的简单的链和密度约束,另一方面,高噪声样本又会因为自身分布外的性质使得在特定数据上训练的针对蛋白体系的神经网络架构性能难以发挥,这可能使得模型会较难学到基本约束并生成不符合常识的序列结构。 本文则回避了这一问题,通过白化去除数据中冗余的相关性关系,并在白化坐标上进行无关扩散,并引入了SO(3)不变性的保留链和密度约束的线形复杂度的多变量高斯分布以实现这些基本约束,这等同于在原始坐标上进行相关扩散。 模型训练使用了以2.6埃,50% identity为阈值筛选得到的28819个pdb以及1726个90% identity过滤的抗体,通过PyRosetta填补缺失,8:2:2根据相似度划分数据集。主链模型在8个GPU训练优化ELBO损失函数,设计网络在1个GPU优化序列和chi角的损失函数。
2.2 低温采样本文首先指出了似然模型的通病,即最大似然拟合的低容量模型通常会过度分散。解决的思路一般为以减少样本多样性为代价增加对高似然状态的采样,比如贪婪、波束采样等方法。低温采样也是其中一种方法,在原本采样分布基础上添加温度项以实现多样性和似然的平衡。 对于扩散模型也存在同样的多样性和质量的平衡,Dhariwal等人[3]表示简单的调整,如对分数函数或噪声的缩放是有问题的,在DALL-E 2、Imagen等模型中往往使用的基于分类器的引导方法。本文提出进行了2个操作并将两者融合使用:1. 对反向扩散的分数函数进行温度项进行缩放,值得注意的是虽然该方法可以起到平衡作用,但不足以在温度扰动的分布中进行适当的重新加权。2. 引入退火Langevin动力学的思想从低温分布中采样,加入平衡率选项,虽然该方法需要有足够的平衡时间。 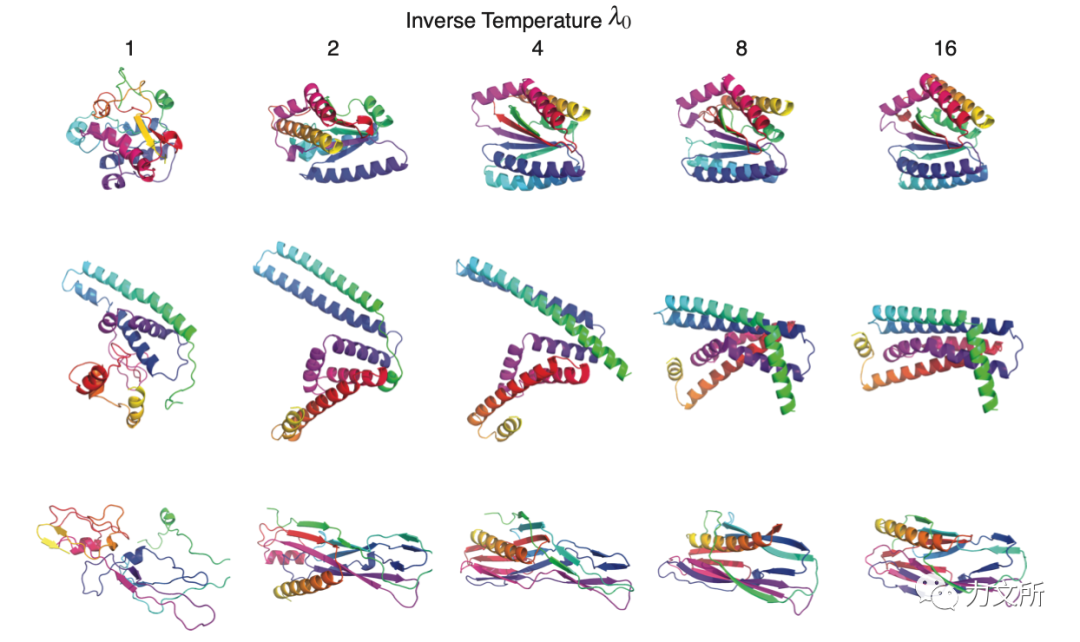
****
**2.3 **随机图神经网络
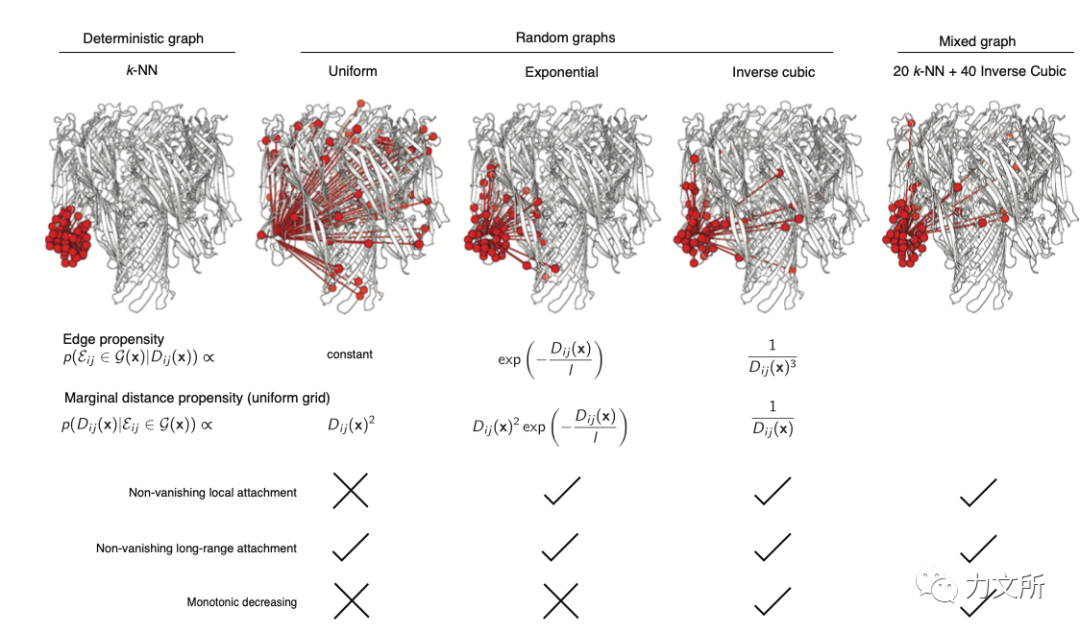
与以往的高复杂度的网络相比,本文复现了高效N-body模拟方法Barnes-Hut算法,并基于消息传递网络和随机图构建了随机图神经网络。这么做的前提之一是计算物理学的主要经验,涉及O(N2)密集相互作用的N-body模拟往往只需要O(NlogN)尺度的计算就可以有效模拟。另一个前提是在transformer的衍生应用中有不少也探索了在降低计算复杂度的前提下加入长距离信息。 目前的文献中,大多数蛋白质结构的图神经网络或者以基于局部连接,例如k-NN,或全连接图的方式处理信息。局部图对于高度空间定位的任务来说是很自然的,比如结构条件下的序列设计和特定残基环境的描述,并且计算负担较小,但是否能够有效地推理全局结构依旧存疑。 本文的方法对短距离和长距离连接混合的随机图进行采样。具体来说,每个节点的边从潜在的边集合中不放回地加权抽样,权重来自于空间距离,最终图使用20近邻边以及40随机逆立方采样边(Barnes-Hut方法)。该方法在于统一采样、指数采样相比,该方法没有丢失局部信息也没有引入额外的长度分布,保留了短距离和长距离的平衡,边际采样概率随距离缓慢单调地衰减。
2.4 坐标更新
在Alphafold的工作中,生成蛋白质几何结构的神经网络对整个分子系统进行了迭代局部坐标更新。本文将该问题类比为机器学习中的 "结构化预测 "问题学习势能函数,并利用优化和几何方法进行解决。关于Alphafold可以学习到势能函数也在Sergey等人[4]的论文中得到证实。本文提出了一种新的凸优化方法实现残基间几何预测和端到端可微调优化,可以将成对的残基间几何信息和置信度信息转换为成对的旋转平移信息,将问题变换为在旋转平移空间内通过局部分析与反复迭代求解全局优化结构的一个凸优化问题。 在上述过程中,局部优化时邻居是固定的,目标函数被定义为:

其中w为置信度,t代表平移,O代表旋转,Proj函数为Kabsch 算法的投影以计算优化RMSD的位置,这个过程被迭代以求得优化后的局部pose,并最终使用Alphafold2中相同的函数再转换为坐标。另外本文也进行了一些局部信息增强的操作。
2.5 可编程性
Chroma的另一个比较大亮点是它的可编程性,可以针对下游任务的需求进行条件性偏向性采样,比如在训练时引入的特定的标签。
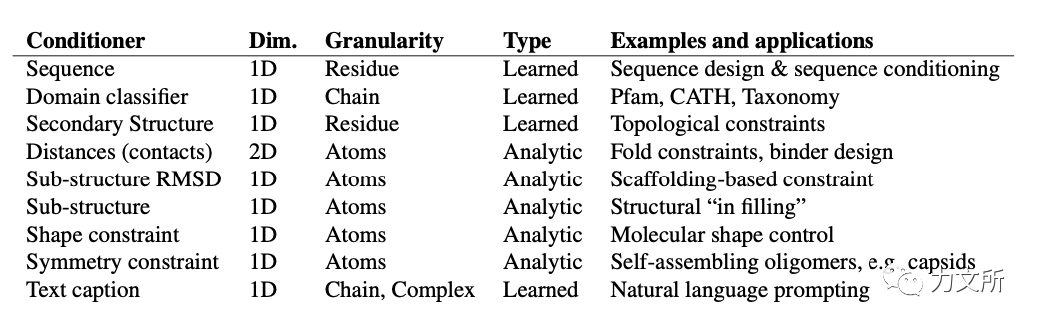
上图展示了本文进行的条件生成的类型,主要包括:1. 距离约束(contact约束)通过指定某些氨基酸对在空间上接近,可以在包括binder设计,环肽设计等场景上应用。操作方式为两种,或者训练一个分类器判别在去噪之后某些位点间距离是否小于某阈值,或者根据扩散过程中的坐标信息直接计算某些位点间的距离以拟合分布。2. 子结构RMSD匹配(motif)这里的应用场景是基于具有潜在功能的特定子结构进行生产,这个特定子结构可以是某结合位点,可以是结合骨架、也可以是特定的表位。操作方式同样分两种,或者为建立一个分类器以评价去噪后的结构与motif之间的RMSA是否小于某阈值,或者同样根据扩散过程计算在不同时间节点的结构匹配RMSD,拟合RMSD变化与噪声和motif之间函数,构建一个RMSD的分类器。3. 子结构填充这里的应用场景为不仅给定了子结构给定,也同样给定了具体的序列位置,即残基编号。此时对给定片段之间的缺失部位的采样变为了双侧节点为缺失部位两侧片段的布朗桥,对分类器的评价依旧是RMSD。另外本文也设置了专门的损失函数避免采样过程中的碰撞和不连续。4. 对称性Chroma同样可以处理对称设计问题,这对于大型蛋白对称组装体很有帮助。同样本文在对可能破坏对称性的错误进行限制后,提出了2种操作方式,一种称为对称广播,计算全局梯度更新并将其和噪音投影到所有对称位置;另一种称为对称平均,平均对称子单元的更新和噪音,这两种方式都可用于更新采样过程并引进对称性。5. 形状除了上述的很贴近应用的一些条件采样之外,作者还探索了一些独特的采样方式。考虑到蛋白质经常通过特定的形状实现特定的功能,比如受体、细胞孔隙等,本文探索了利用形状约束对蛋白质进行采样。约束方法来自最优传输方法[5] ,这种方法可用于比较残基之间的对应关系和几何距离。通过对预测距离矩阵和理想形状的距离矩阵计算Wasserstein距离(点云距离)和Gromov- Wasserstein距离(不同domain之间的未对齐距离),添加形状的损失函数用于优化采样。6. 残基或者更高层次上的分类模型
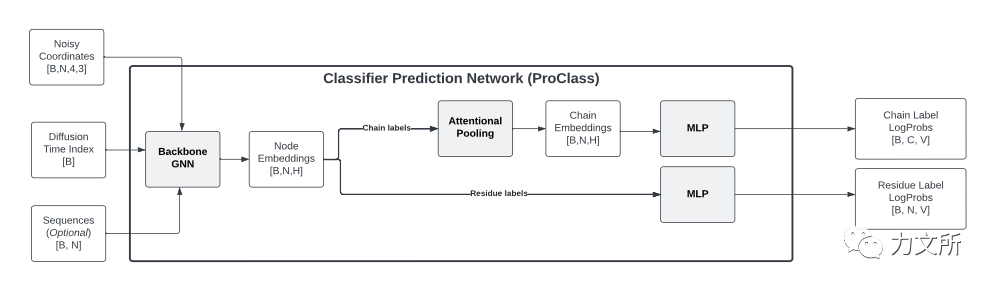
在以往的方法中对于特定领域的迁移往往使用微调或者自适应迁移等方法。本文则另训练了一个模型ProClass对于加噪的主链坐标信息进行分类,该模型的架构如上所示,训练数据为CATH、PFAM、二级结构、界面残基等等信息,这意味这扩散模型也可以根据该分类器向着特定二级结构,界面残基,家族进行生成。7. 自然语言
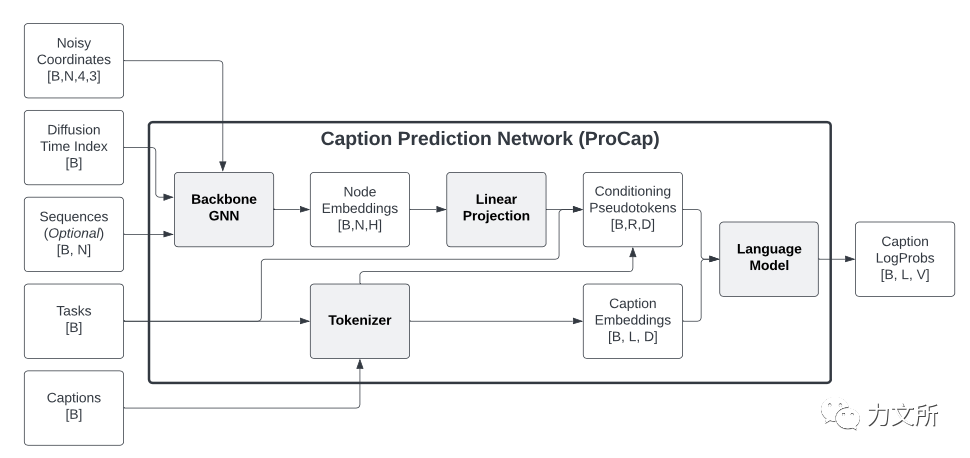
本文最新颖的案例则为仿照AIGC的方法,从特定的自然语言提示出发进行蛋白生成。本文训练了一个从文本到蛋白质骨架的生成模型ProCap,该模型使用在科学文本上训练的1.25亿参数模型GPT-Neo,期望它能够理解和蛋白质相关的语言描述,也使用了ProClass的网络以编码主链。模型的输入包括由ProClass编码的结构和由GPT-Neo编码的语言描述和任务字符,这里的任务字符用于区分是单链任务还是复合物任务。如上图架构所示,ProCap在语言和结构模型外,通过可学习的线性层来连接这两个模型,一个可学习的语言模型头来将原始语言模型输出转换为概率。 ProCap在约45000个由PDB和Uniprot组成的文本描述-结构的数据对上使用单张V100训练,其中的文本描述来自网站上的描述性文本或者注释。
**
**
**
**
**3. **结果分析
在结果展示部分,文章依次对扩散非条件生成和各种可编程性的定向生成结果进行了基于拆分数据集的分析。生成过程效率很高,只需要使用一般GPU,对于3000个氨基酸以上的复合物生成只需要几分钟。
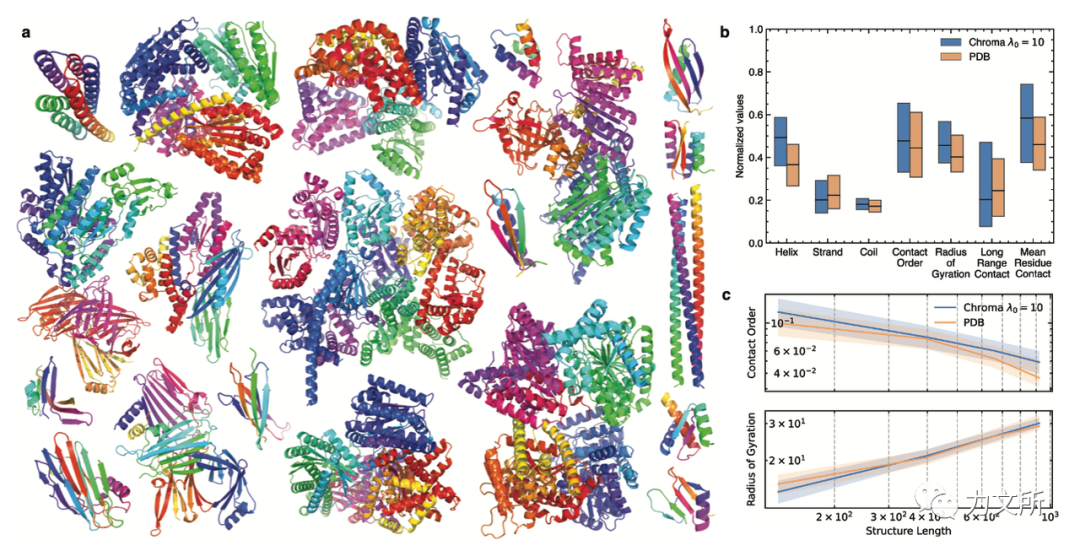
首先在非条件生成上,作者比较了低温采样带来的影响,如果没有低温采样,模型会天然结构分配了很高的似然,同时生成蛋白大多为不合理的结构,比如有很少的二级结构。而加入了低温采样则会缓解该问题,图左侧展示了大量生成出的结构,图右侧则对一些单链性质,比如二级结构分布、contact密度、长距离contact,回转半径等进行了生成蛋白和和天然蛋白的比较,两者的相似度很高,生成蛋白的螺旋比较多,这一定程度上说明生成结果的合理性。 另一方面本文也对生成结果进行了Omegafold评价、与天然TERMs的比对分析均显示了较高的可设计性。
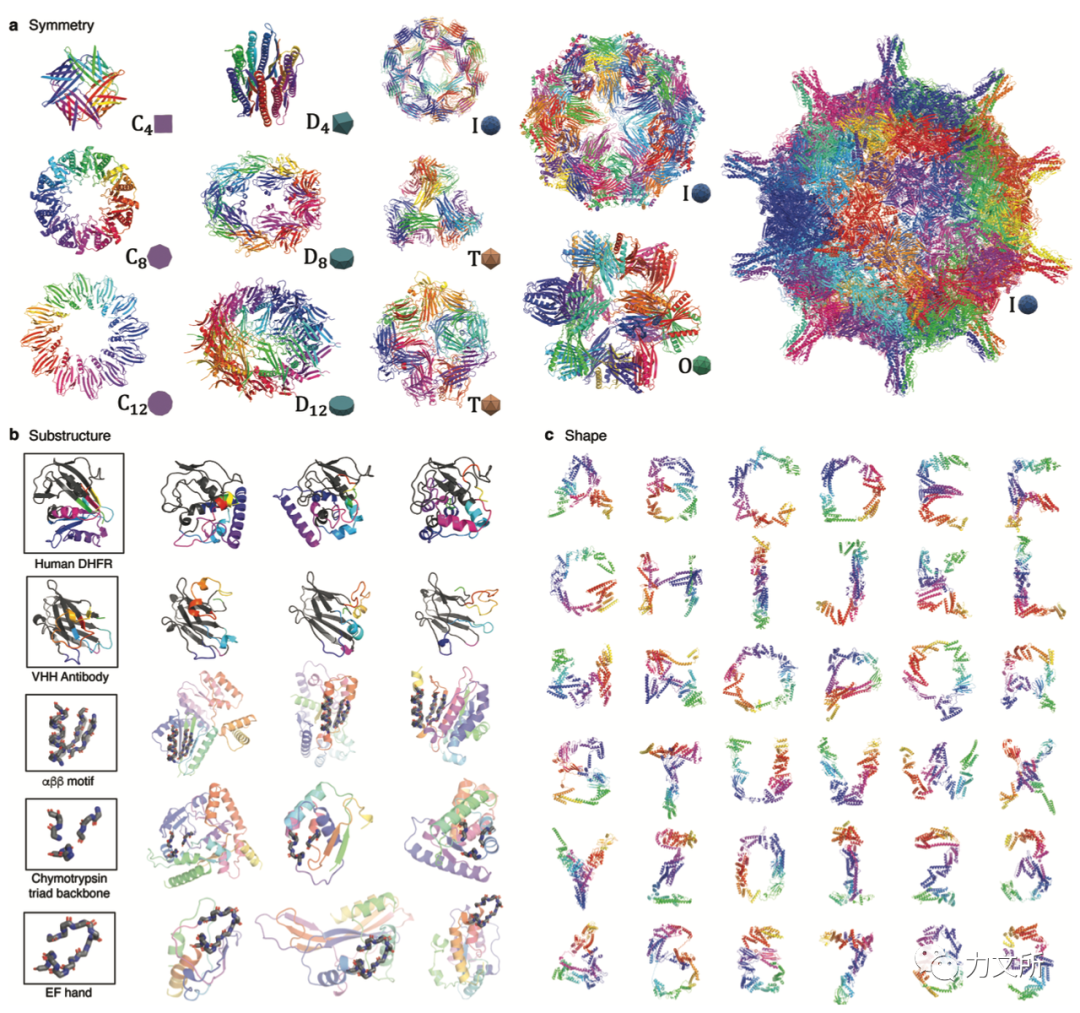
上图展示了“可编程性”的一部分结果。1. 几何约束(a图):展示了对称性采样,上图展示了更多的对称生成样本,包括各种的循环对称、二面体对称、四面体对称、八面体对称和二十面体对称。这样的对称性设置很严格,作者发现甚至有一些对称的样本中间出现了空腔,不具备良好的接触界面,这也是后续可能需要改进的问题。受益于本文的高效计算,这里展示的最大案例为包含96000个残基的二十聚体。2. 子结构(b图):本文展示了5个案例,包括对二氢叶酸还原酶一半结构的重构,抗体对CDR的重构,其他预先指定的motif(αββ packing motif,编码糜蛋白酶催化三联体活性部位的骨架片段,EF-hand Ca-binding motif)的自由生成,结果显示可以生成具备多样性的合理的结构。3. 形状(c图),本文以英文字母和阿拉伯数字作为形状约束进行生成,从结果来看虽然无法评价合理性,但是在二级结构,侧链packing方面都较为正常。
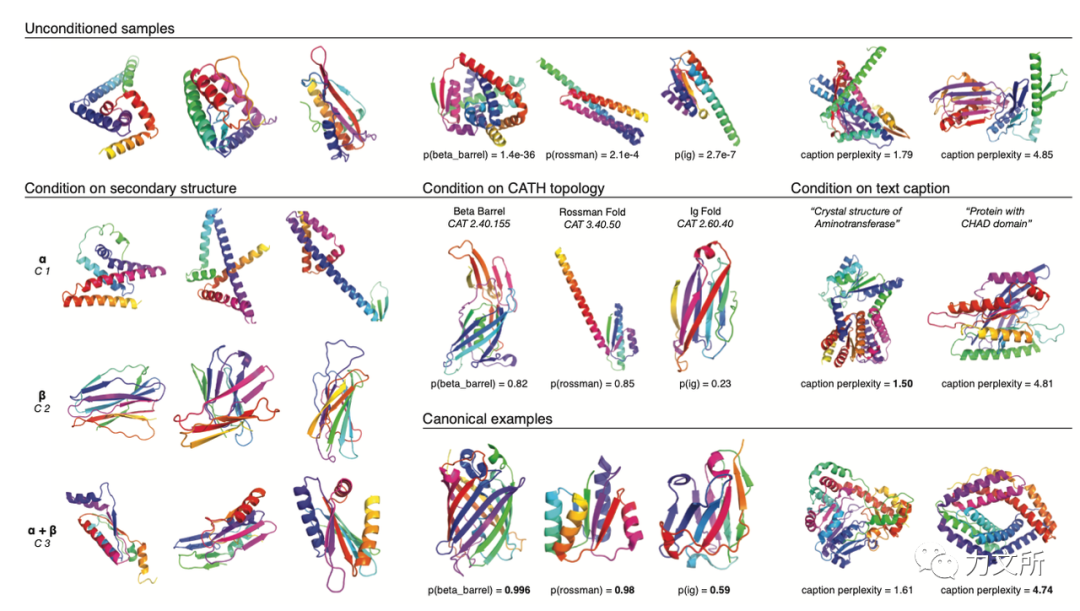
上图展示了通过ProClass和ProCap进行条件生成的结果,所有竖行之间都共享相同的随机种子有对应关系,可以看到在二级结构方面有着肉眼可见的对特定二级结构组合的富集,在折叠类型的生成上对于beta桶和Ig折叠的生成效果较好,但是对于Rossman折叠上则只具备一些特征,出现了和非条件生成一样的很长的单螺旋。 右侧则展示了自然语言条件的生成结果,可以看到条件生成结果使得模型的困惑度的到了提升,甚至在一个案例上超越了PDB结构。另外作者也在案例分析时指出,语言模型条件并不是经常生效的,一个影响很大的因素是语言模型的梯度和结构扩散模型梯度的融合,语言模型梯度权重过小会导致和无条件生成没有区别,大了又会导致生成样本质量下降,这些都是后续需要讨论的问题。 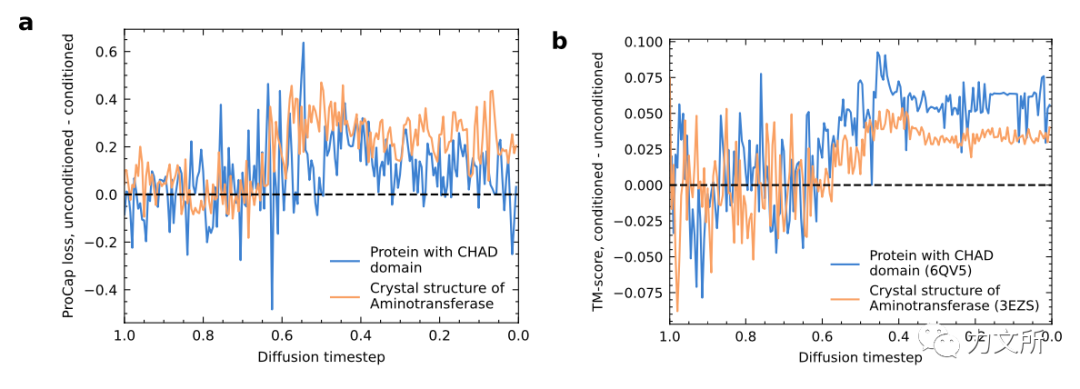
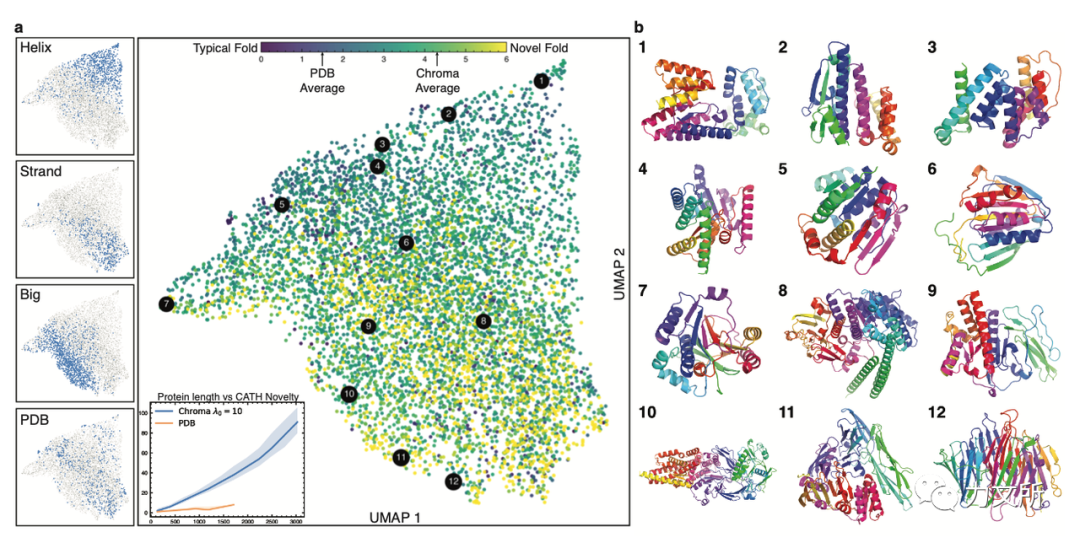
最后则是对整个蛋白空间进行可视化,对应标题照亮蛋白空间。本文与之前蛋白空间可视化的语言模型相比,有着明确的结构信息并可以将这些结构信息与空间位置进行严格对应,正如左侧展示的一样,相似的类别二级结构比例,大小,在空间中产生了明显聚集。 这里引入了一个新颖度评价方式“CATH-可压缩性”,即在TM分数高于0.5的情况下,实现对蛋白质中80%的残基的贪婪覆盖所需的CATH域的数量,作者意图说明生成蛋白在新颖度上有着明显的优势且具备多样性,一方面,左下角的子图中展示了天然pdb和Chroma生成蛋白随着长度增加生成蛋白的新颖度变化,生成蛋白占据明显优势。另一方面,本文将蛋白的拓扑描述符进行Umap投影,并根据新颖度进行上色,以右侧的12个为代表的生成蛋白不仅覆盖广泛,而且有着极高的新颖度。
4. 力评
Chroma作为一个新扩散生成模型,在扩散模型、AIGC和Alphafold2正值风口的当下,与bakerlab的RFdiffusion同期发布。它吸纳了众多模型的优点实现了高效高质量的可编程蛋白设计,同时文章也指出了一些可以调整的设置作为开放问题,这会进一步促使AI蛋白设计领域的发展。该模型潜在应用广泛,包括邦德设计、以抗原做条件的抗体设计、酶的活性位点嫁接、材料设计等等,这些应用都亟待改进以及实验验证。 Chroma 旨在转移蛋白设计的焦点问题,将主链采样问题转向特定任务特定功能的采样——即蛋白质打算做什么。通过利用大自然积累的数以亿计的蛋白质进化实验,Chroma 等生成模型有望推动人类健康和生物工程的生物分子多样性的另一次改变。
参考链接: 1. Illuminating protein space with a programmable generative model. John Ingraham, Max Baranov, Zak Costello, Vincent Frappier, Ahmed Ismail, Shan Tie, Wujie Wang, Vincent Xue, Fritz Obermeyer, Andrew Beam, Gevorg Grigoryan. bioRxiv 2022.12.01.518682 1. John Ingraham, Vikas Garg, Regina Barzilay, and Tommi Jaakkola. Generative models for graph- based protein design. Advances in neural information processing systems, 32, 2019. 1. Namrata Anand, Raphael Eguchi, Irimpan I Mathews, Carla P Perez, Alexander Derry, Russ B Altman, and Po-Ssu Huang. Protein sequence design with a learned potential. Nature commu- nications, 13(1):1–11, 2022. 1. Prafulla Dhariwal and Alexander Nichol. Diffusion models beat gans on image synthesis. Ad- vances in Neural Information Processing Systems, 34:8780–8794, 2021. 1. State-of-the-art estimation of protein model accuracy using AlphaFold. James P. Roney, Sergey Ovchinnikov. bioRxiv 2022.03.11.484043 1. Gabriel Peyre ́, Marco Cuturi, et al. Computational optimal transport: With applications to data science. Foundations and Trends® in Machine Learning, 11(5-6):355–607, 2019.
THE END

如对蛋白设计有合作兴趣或者疑惑? 请发送邮件至:info@levinthal.bio 联系人:雷经理 13166462262 详情请查询官网:http://levinthal.bio/ ☻ ©️ 力文所原创内容,未经许可转载必究。