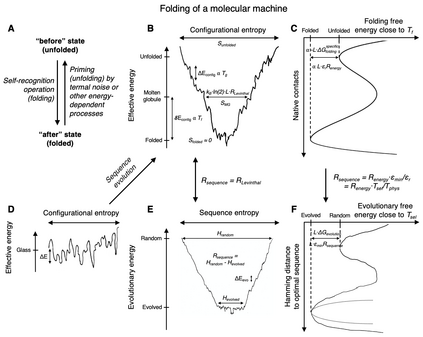
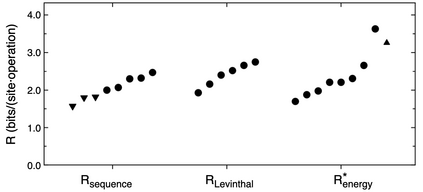
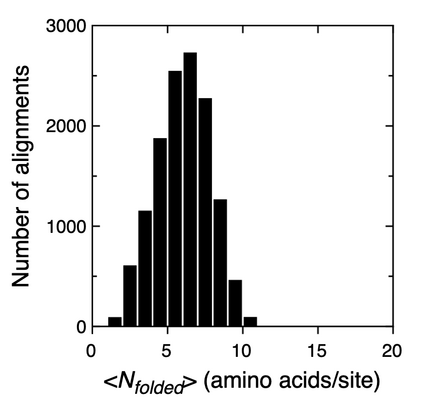
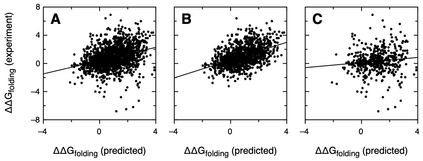
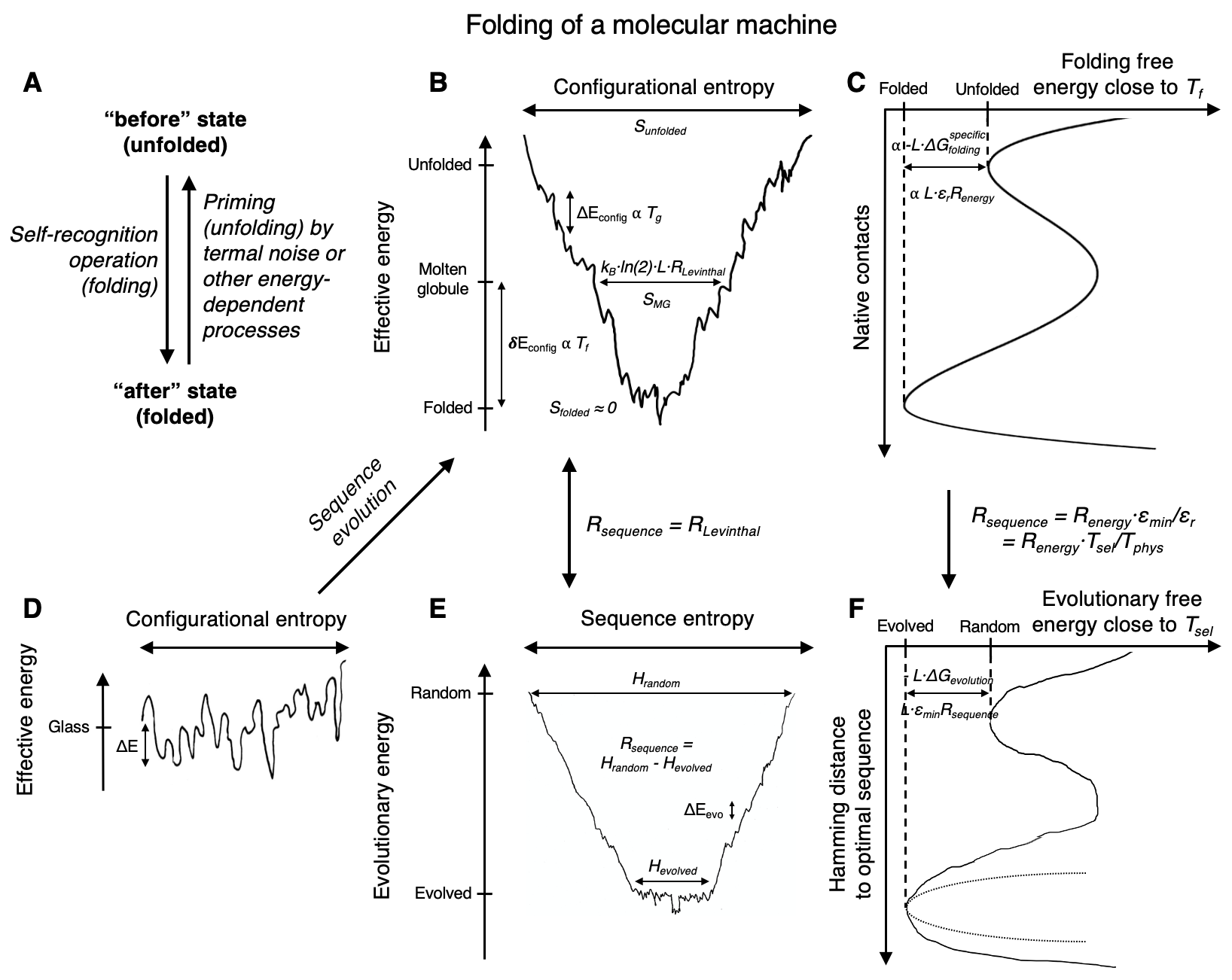
We propose an application of molecular information theory to analyze the folding of single domain proteins. We analyze results from various areas of protein science, such as sequence-based potentials, reduced amino acid alphabets, backbone configurational entropy, secondary structure content, residue burial layers, and mutational studies of protein stability changes. We found that the average information contained in the sequences of evolved proteins is very close to the average information needed to specify a fold ~2.2 $\pm$ 0.3 bits/(site operation). The effective alphabet size in evolved proteins equals the effective number of conformations of a residue in the compact unfolded state at around 5. We calculated an energy-to-information conversion efficiency upon folding of around 50%, lower than the theoretical limit of 70%, but much higher than human built macroscopic machines. We propose a simple mapping between molecular information theory and energy landscape theory and explore the connections between sequence evolution, configurational entropy and the energetics of protein folding.
翻译:我们建议应用分子信息理论来分析单领域蛋白质的折叠情况。 我们分析来自蛋白质科学各个领域的结果, 如基于序列的潜力, 减少氨基酸字母, 脊椎配置酶, 二次结构内容, 残余埋层, 以及蛋白质稳定性变化的突变研究。 我们发现进化蛋白序列中包含的平均信息非常接近指定一个折叠 ~ 2.2 $\ pm$ 0.3 比特/(现场操作) 所需的平均信息。 进化蛋白中的有效字母大小等于5 左右缩放状态中残留物的有效相容数量。 我们计算了在折叠上大约50%的能量到信息转换效率, 低于70%的理论限制, 但远高于人造的宏观机器。 我们建议对分子信息理论和能源景观理论进行简单的绘图, 并探索序列进化、 配置酶 和蛋白折合能之间的关联。