作者 | 郑仰昆 审稿 | 杨崇周 指导 | 闵小平(厦门大学) 今天带来的是美国马萨诸塞州波士顿哈佛医学院系统药理学实验室发表在nature biotechnology上的Single-sequence protein structure prediction using a language model and deep learning。

单序列结构预测是较为基础的研究方向,蛋白质设计和量化序列变异对功能或免疫原性影响的研究等都需要单序列结构预测作为支持。AlphaFold2 和相关计算系统使用以多序列比对 (MSA) 编码的深度学习和共同进化关系来预测蛋白质结构。尽管这些系统有很高的预测准确性,但其对于无法生成 MSA 的孤儿蛋白质的预测、快速设计结构仍然有些不足。
本文针对以上两个问题设计了一个端到端可微循环几何网络 (RGN2),该网络使用蛋白质语言模型 (AminoBERT) 从未对齐的蛋白质中学习潜在的结构信息,以此改进之前提出的RGN。RGN2 在孤儿蛋白质和设计蛋白质类别上的性能优于 AlphaFold2 和 RoseTTAFold,同时计算时间减少了 106 倍。并证明了蛋白质语言模型在结构预测中相对于 MSA 的实践和理论优势。
模型构造
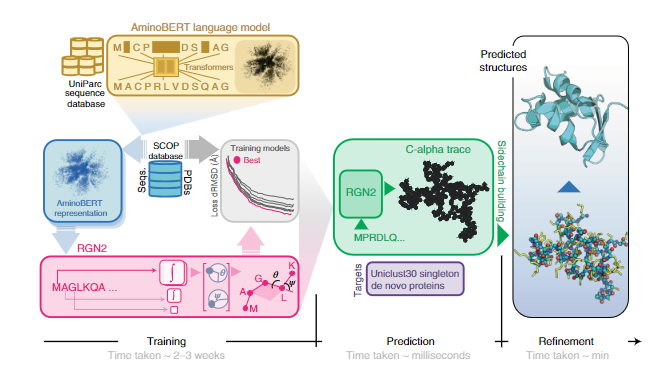
图1 RGN2的组织与应用
RGN2组成:RGN2 将基于转换器的蛋白质语言模型(AminoBERT,黄色)与使用 Frenet-Serret 框架生成蛋白质骨架结构(绿色)的 RGN 相结合。在初步构建侧链和氢键网络后,随后使用 AF2Rank(蓝色)对结构进行细化。
**RGN:**基于机器学习的 RGN,利用源自 MSA 的位置特异性评分矩阵(PSSM)预测蛋白质结构,将PSSM 结构关系参数化为相邻残基之间的扭转角,从而可以在 3D 空间中顺序定位蛋白质骨架(骨架几何结构包括每个氨基酸的 N、Cα 和 C' 原子的排列)。尽管 RGN1 不依赖用于生成 MSA 的协同进化信息,但对 PSSM 的要求需要多个同源序列可用。RGN2改进了RGN,利用了一种自然的方式来描述在整个多肽水平上旋转和平移不变的多肽几何形状。这涉及使用 Frenet-Serret 公式在每个 Cα 碳嵌入参考框架;然后通过一系列转换轻松构建主干。
**AminoBERT: **AminoBERT 旨在捕获一串隐含指定蛋白质结构的氨基酸中的潜在信息。为了生成 AminoBERT 语言模型,本文使用从 UniParc 序列数据库获得的约 2.5 亿天然蛋白质序列训练了一个 12 层转换器。训练任务第一个是预测序列中同时屏蔽的2-8个连续残基,强调从全局而不是局部上下文中学习。第二个是识别打乱的“块排列”顺序,块排列是连续的蛋白质片段交换,保留了局部序列信息,但破坏了全局连贯性,鼓励转换器从整个蛋白质序列中发现信息。RGN2 的 AminoBERT 模块以自我监督的方式独立于几何模块进行训练,无需微调。
**数据:**RGN2 训练是使用 ProteinNet12 数据集和仅由源自 ASTRAL SCOPe 数据集(版本 1.75)的单个蛋白质域组成的较小数据集进行的。因为本文观察到两者之间没有可检测到的差异。
图2比较 RGN2 和 AF2 对孤儿蛋白的结构预测
表1 RGN2 和 AF2、RF 和 trRosetta 跨 330 个目标的预测时间比较
**结果:**本文使用dRMSD 和 GDT_TS 评估了预测准确性。堆积条形图2显示了 149 种从头设计的孤儿蛋白质。条形高度表示蛋白质长度。对于富含单螺旋和弯曲或散布有螺旋的氢键转角的蛋白质,RGN2 优于所有其他方法。表1展示了对于没有同源序列的蛋白预测时花费的时间是RGN2明显占优的。
总结
RGN2 是使用机器学习从单个序列预测蛋白质结构的首次尝试之一。在设计孤儿蛋白质结构的情况下具有许多优势,因为这些蛋白质通常无法生成多序列比对。RGN2 通过将蛋白质语言模型 (AminoBERT) 与基于 Frenet-Serret 公式的简单直观的 Cα 骨架几何参数化方法融合来实现这一点。AF2 和 RF 的无模板和无 MSA 生成均比 RGN2 慢 >105 倍。本文认为,未来同时使用语言模型和 MSA 的混合方法可能会优于单独使用任何一种方法。
参考资料 Chowdhury, R., Bouatta, N., Biswas, S. et al. Single-sequence protein structure prediction using a language model and deep learning. Nat Biotechnol (2022). https://doi.org/10.1038/s41587-022-01432-w