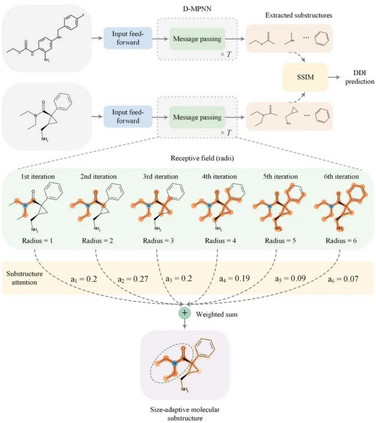
2022年7月13日,中山大学陈语谦团队在Chemical Science上发表文章。作者提出了一种子结构感知图神经网络,以学习尺度自适应的药物分子关键子结构,从而对药物-药物相关性进行可解释性预测(Learning size-adaptive molecular substructures for explainable drug–drug interaction prediction by substructure-aware graph neural network,SA-DDI)。

该网络结合了新的子结构注意机制和用于DDI预测的子结构-子结构交互模块(substructure-substructure interaction module,SSIM)的消息传递神经网络。 具体而言,子结构注意力是基于分子中官能团的尺寸和形状通常不规则的化学直觉,设计来捕捉尺寸和形状自适应的子结构。DDI基本上是由化学子结构相互作用引起的。因此,**SSIM通过突出重要的子结构,而不强调次要的子结构用于DDI预测,从而对子结构-子结构相互作用进行建模。**SA-DDI超过了其他方法,且SA-DDI对药物的结构信息敏感,能够检测DDI的关键子结构。这些优点表明,该方法提高了DDI预测建模的泛化和解释能力。
背景
药物-药物相互作用(drug-drug interaction,DDI)会对人体产生难以预期的药理作用,其因果机制通常未知。图神经网络(GNN)已被开发用于更好地理解DDI。然而,识别对DDI预测贡献最大的关键子结构对GNN来说仍是一个挑战。因为,GNN最常见的读出函数(即全局平均值或者加和池化)不适合DDI预测。例如,通过直接计算子结构表示的总和或者平均值,主要的子结构可能被次要的子结构信息掩盖。所以,有必要引入注意力机制与挖掘药物相互感知作用的策略,自适应地捕捉药物分子的子结构,更好地进行DDI预测。 方法
SA-DDI总体框架如图1所示。一般而言,DDI预测任务是开发一个计算模型,该模型将两种药物作为输入,并生成一个输出预测,指示它们之间是否存在相互作用(即副作用)。首先,利用输入前馈模块(即多层感知器)对节点进行非线性变换,以获得更好的特征表示。然后,将两个分子图输入到配备有子结构注意力的GNN中,以提取尺寸和形状自适应子结构。最后,将提取的子结构输入SSIM,以学习子结构-子结构相互作用,模型从中进行DDI预测。 GNN因其在描述分子的原子和键的化学问题中的自然适用性而受到关注。一般来说,GNN由以下三个阶段组成:(1)通过聚合来自其邻居节点的消息(即消息传递)来更新节点级特征;(2)通过使用读出函数聚合分子图中的所有节点级特征,生成图级特征向量;(3)基于图级特征向量预测图的标签。 在第一阶段,节点级隐藏特征通过在相邻节点之间传递消息来更新T次(即T次迭代)。在每次迭代中,代表节点半径的感受野可以通过访问其相邻节点的信息来放大。节点可以被视为在第T次迭代后以自身为中心、半径为T的子结构。然后,在最后一个时间步骤T处更新的节点级隐藏特征在所有节点上聚合,以生成给定图的图级特征向量。最后,使用图级特征向量来预测整个图的标签,例如分子性质。 在这项研究中,作者使用有向图消息传递神经网络(directed message passing neural networks,D-MPNN),用于分子子结构提取。然而在第二阶段中,典型的读出函数计算来自图的所有节点级特征的平均值或者总和,以获得给定图的图级表示,但它对于DDI预测具有缺点。因此,作者引入了新的子结构注意机制,也就是通过自注意力池化(self-attention graph pooling,SAGPool)读出函数计算给定图的图级表示。
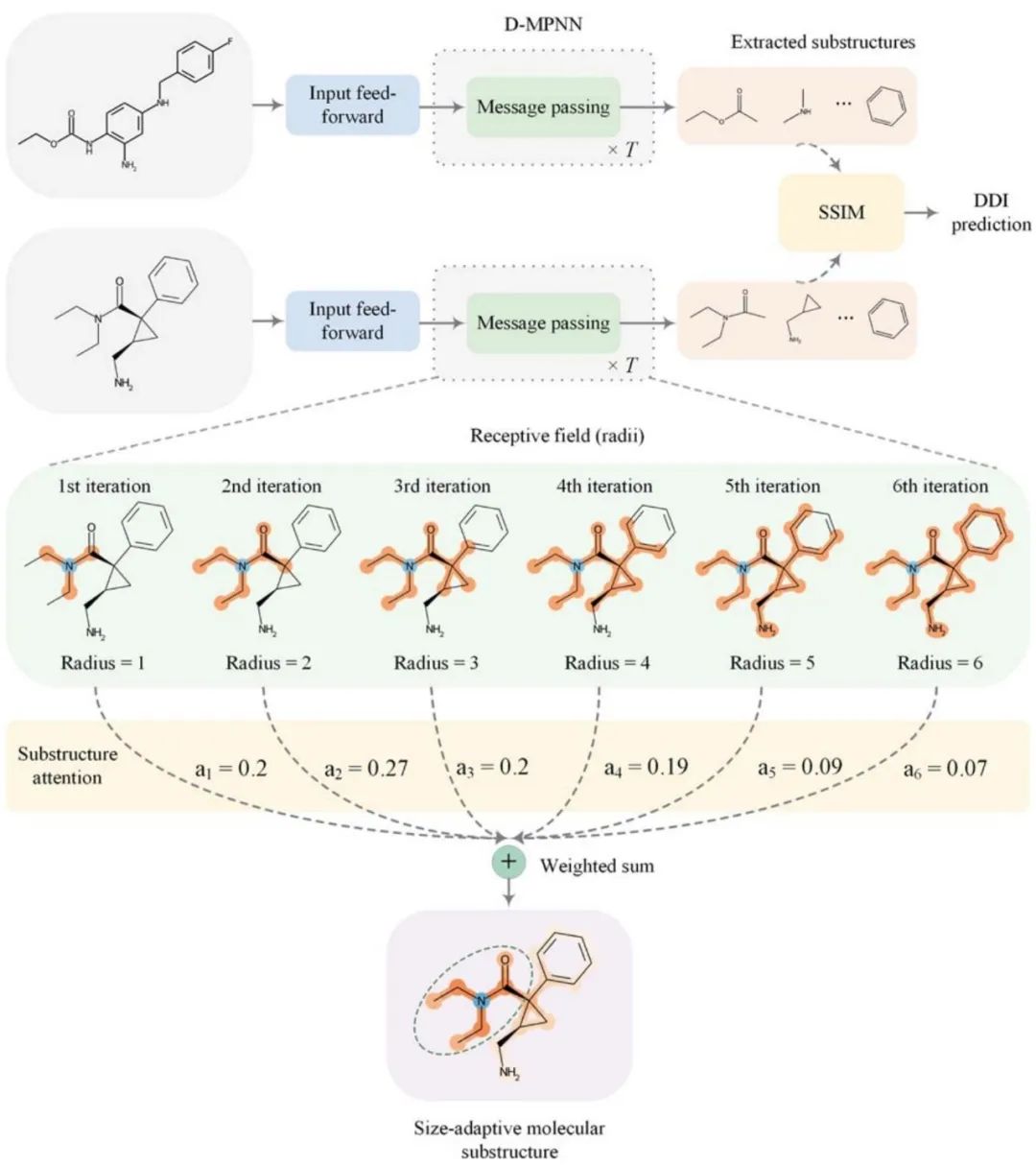
图1. SA-DDI模型 在使用SAGPool得到分子图的初步表示之后,子结构-子结构相互作用模块(SSIM)通过交互一对药物分子的图级别表示和节点级别表示信息,更新分子图的表示,通过拼接一对药物的表示向量并输入多层感知器得到最终预测结果(是否具有相互作用)。
结果
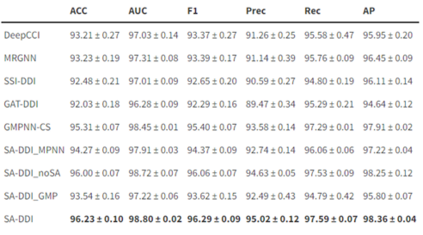
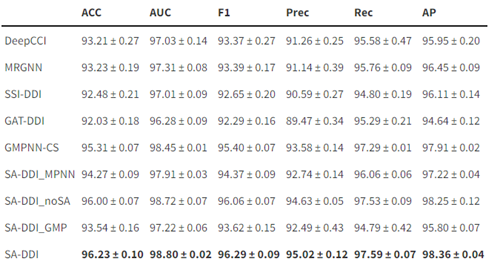
作者将所提出的SA-DDI与现有的多种方法进行了比较。为了研究D-MPNN,子结构注意力和子结构-子结构交互模块如何提高模型性能,还考虑SA-DDI的以下变体: SA-DDI_MPNN将D-MPNN替换为MPNN。 SA-DDI_noSA是SA-DDI的一种变体,可消除子结构注意力。 SA-DDI_GMP用全局平均池化代替SSIM。 表1表明,使用D-MPNN,子结构注意力和子结构-子结构交互模块的SA-DDI,在各项指标上均超越了其他方法。 表1. 不同方法对比
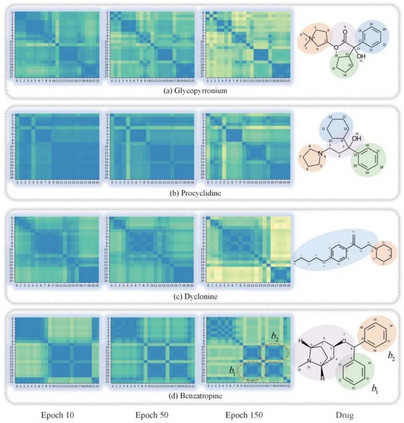
如果不了解和验证GNN的内部工作机制,就不能完全信任GNN,这限制了其在药物发现场景中的应用。为了研究原子节点隐藏向量在学习过程中的演变,作者通过测量图神经网络最后一层的节点隐藏向量的皮尔逊相关系数来获得原子对之间的相似系数。 图2给出了四种药物及其在学习过程中的原子相似度矩阵。热图在开始时显示出某种程度的混乱,然后在学习过程中清晰地分组成簇。以图2(b)普环啶(procyclidine)为例,发现在迭代150次时,原子大致分为四个簇:异丙醇,四氢吡咯,苯基环己烷和苯。这一发现符合我们对其结构的直觉。 这些结果表明,SA-DDI可以捕获分子的结构信息。此外,SA-DDI能够识别分子中的相同官能团,如(d)苄托品(benzatropine)中的苯b1和b2,它还可以区分具有细微结构差异的官能团,如苯基环己烷和苯,如(b)所示。
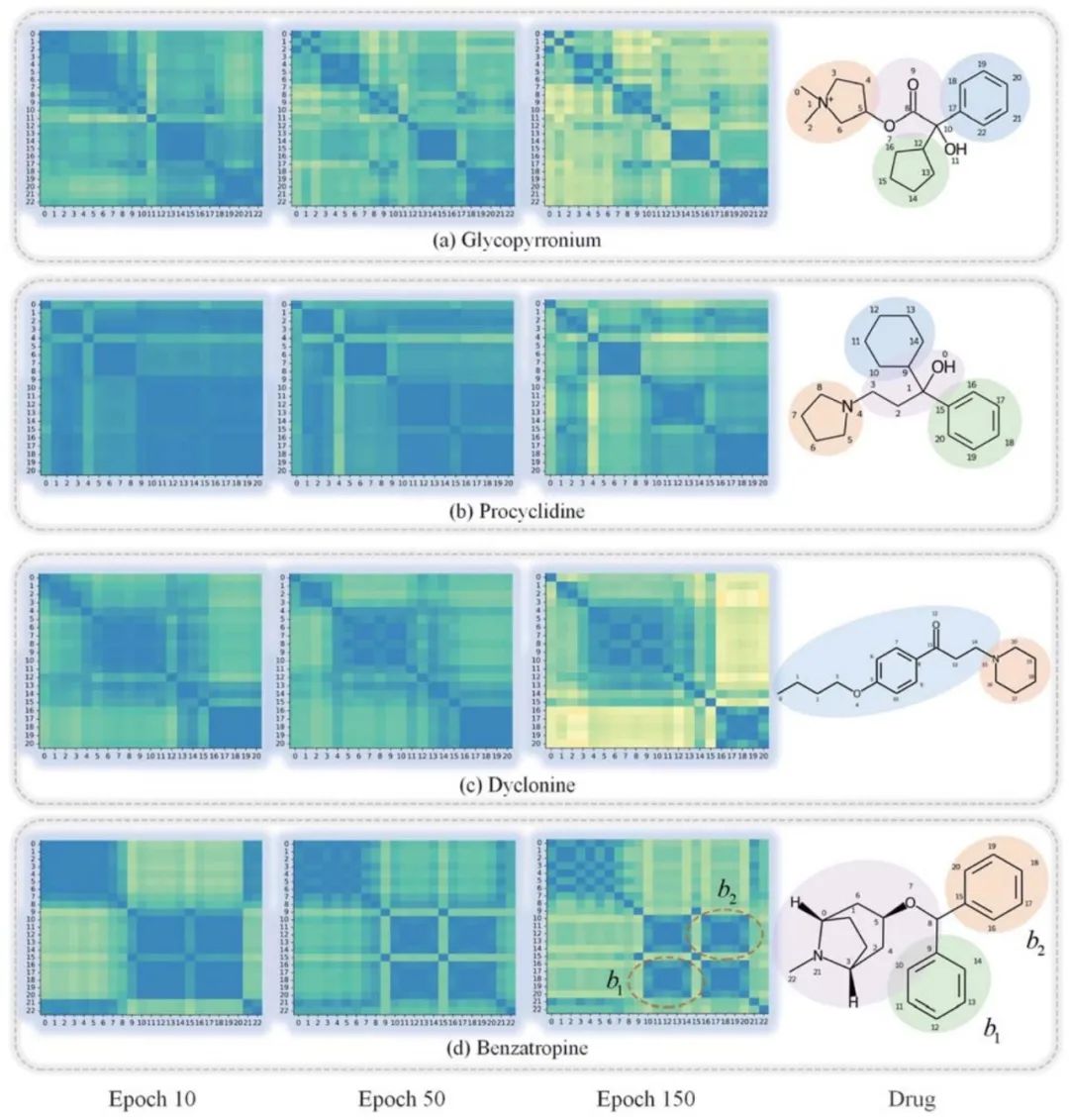
图2. 可视化 总结
本文提出了一种基于图的模型,称为SA-DDI,用于DDI预测。基于DDI从根本上是由化学子结构相互作用引起,本文提出了两种新的策略,包括子结构注意力和相互感知,专门用于检测具有不规则大小和形状的药物分子的子结构,并对子结构与子结构之间的相互作用进行建模。SA-DDI超过了最先进的方法,可以捕获药物的结构信息,并检测DDI的基本子结构,使模型的学习过程更加透明和可操作。SA-DDI是提高DDI预测建模泛化和解释能力的有力工具。 参考资料 [1]Yang et al. Learning size-adaptive molecular substructures for explainable drug–drug interaction prediction by substructure-aware graph neural network. Chem. Sci. 2022 [2]Yang et al. Analyzing Learned Molecular Representations for Property Prediction. J. Chem. Inf. Model. 2019 [3]Lee et al. Self-Attention Graph Pooling. ICML. 2019
--------- End ---------