了解蛋白质序列和功能之间的关系对于设计在生物能源,医学和农业中应用的新颖和有用的蛋白质是十分必要的。从序列到功能的映射非常复杂,涉及数千种在多个长度和时间尺度上耦合的分子相互作用,因此精确预测序列变化对蛋白质的行为和性质的影响是一个重要的问题。Gelman等研究人员提出了一个监督式深度学习框架[1],从深度突变扫描数据中学习序列-功能映射,并对新的、未表征的序列变异进行预测。通过测试多种神经网络架构探索其学习序列-功能映射的能力。研究发现相比基于物理和无监督的预测方法,监督学习方法具有卓越的性能。此外,作者展示了模型探索序列空间和设计超越训练集的新蛋白质的能力。背景
蛋白质序列到功能的映射对于描述自然进化过程,诊断遗传疾病以及设计具有特殊性质的新蛋白质是非常重要的。这种映射由数千种复杂的分子相互作用,动态构象集合以及生物物理性质之间的非线性关系形成。这些高度复杂的特征使得建模和预测氨基酸序列的变化如何影响功能变得具有挑战性。 随着DNA测序、三维结构测定和高通量筛选的进步,蛋白质的序列与结构的数据量呈爆炸式增长。统计学和机器学习方法已成为理解从蛋白质序列到功能的复杂映射的强大方法。无监督学习方法,如EVmutation[2]和DeepSequence[3]通过比对进化相关蛋白质序列,利用大量已联配的进化相关序列进行训练。这些方法可以对蛋白质家族的天然功能作出建模,但无法预测不受长期进化选择影响的特定蛋白质的性质。相比之下有监督方法能直接从序列功能样本中学习到特定蛋白质属性的映射。先前的监督学习方法存在一定局限,如无法捕获蛋白序列到蛋白性质的非线相互作用,对大型数据集的可扩展性差,仅可对单突变变异进行预测等。因此,目前需要通用的、易于使用的监督学习方法,利用大型序列功能数据集来预测特定的分子表型,并满足蛋白质设计所需的高精度。深度学习框架
深度突变扫描数据由数千至数百万个蛋白质序列变体组成,每个变体都有一个对应的评分,可用于量化它们在高通量功能测定中的活性或适应性。通过对蛋白序列进行编码,捕获每个氨基酸的生化及物化性质。训练一个神经网络将编码序列映射到其相关的功能分数后,网络即可被推广为预测新蛋白质变体的功能评分(图1A)。作者测试了四个监督学习模型(线性回归和全连接,序列卷积和图卷积神经网络)的性能,图1D是图卷积网络中的蛋白结构生成过程;结果表明具有参数共享的卷积神经网络能够学习跨不同序列位置的更高层次的特征。
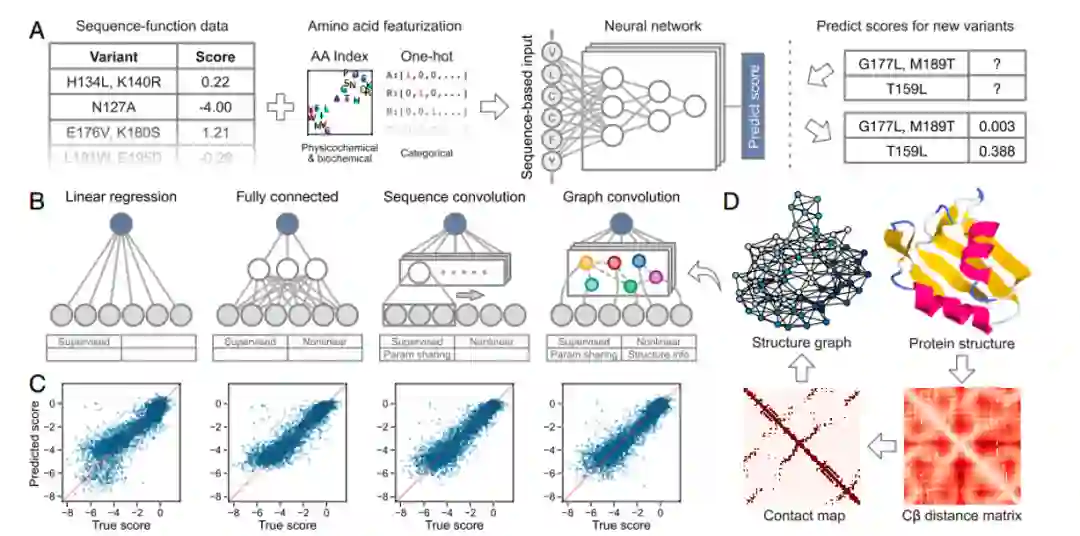
1.模型评估
作者评估了不同网络架构在五个不同(不同大小、折叠和功能的蛋白质,表1)数据集上的预测性能(图2A)。发现模型对大多数据集都表现了出色的预测性能,Pearson相关系数范围为0.55~0.98(图2B)。对大多数蛋白质,卷积网络在较小的训练集上也具有不错的表现(图2C),其在一个数据集上实现了强大的性能(r > 0.9),在另外两个数据集上实现了中等性能(r > 0.6),并在所有数据集中的表现优于线性回归和全连接网络(图2D)。对每个模型进行实验测试(取决于合成和评估给定蛋白质变体的实验成本),评估结果表明监督模型始终比Rosetta和无监督方法实现更高的召回率。(图2E)。
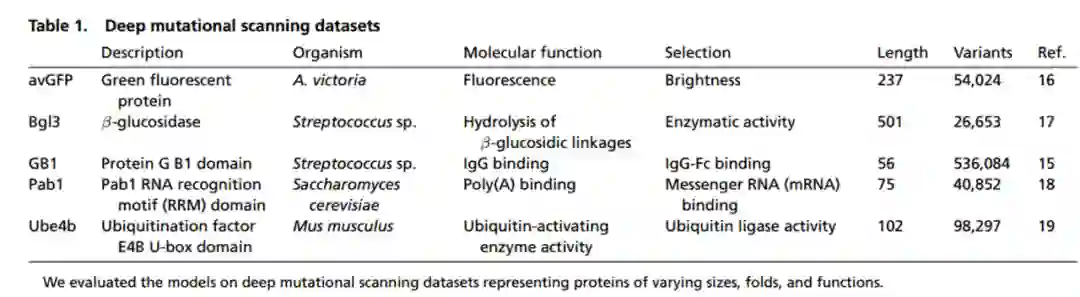
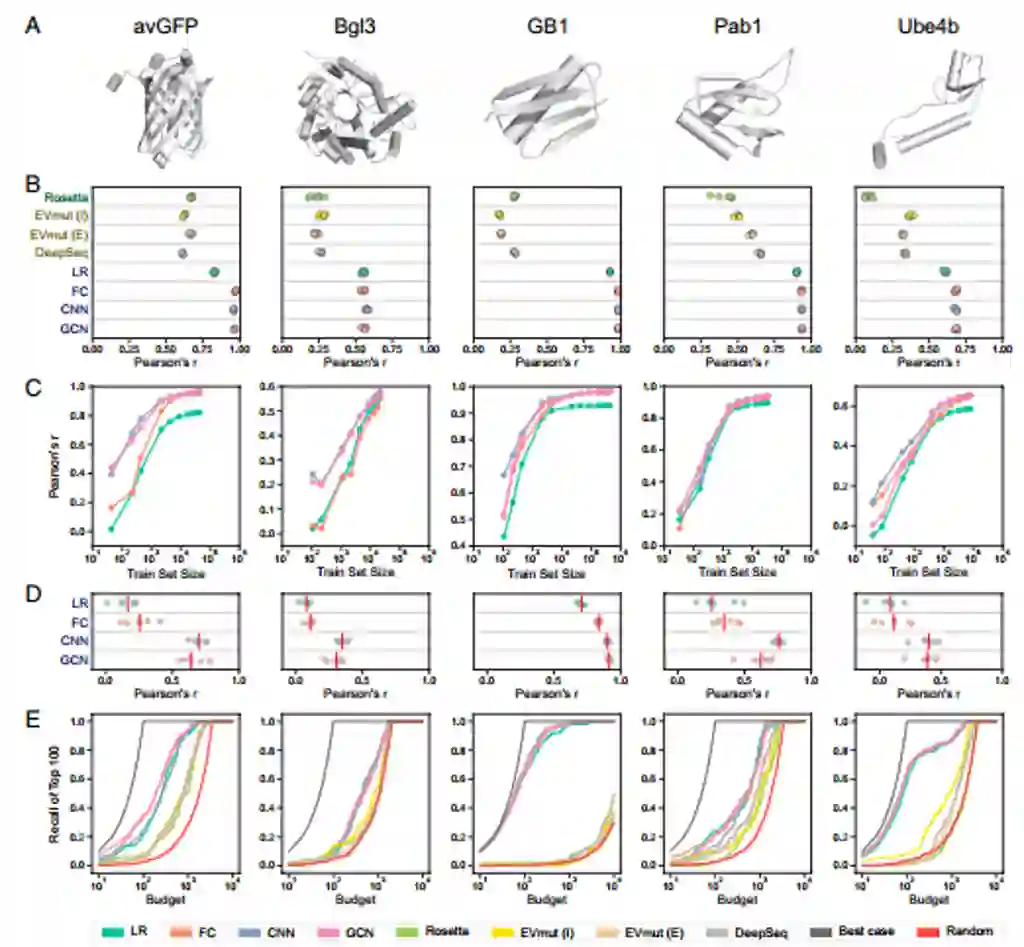
图2 不同模型间预测结果比较**********[1]**********
**2.**数据质量在学习序列功能模型中的作用
作者在每个模拟数据集上训练序列卷积模型,并在“真实”的非重采样测试集上测试了每个网络的预测能力(图3)。结果表明在较小的数据集上训练的模型表现不佳,原因是没有足够的样本来学习序列-功能映射。此结果与在原始GB1数据集上利用减小规模的训练集训练时模型表现一致(图2C)。另外如果没有足够的DNA测序读数来可靠地估计每个变体的出现频率,大数据集训练的性能可能也会很差。因此未来的深度突变扫描文库可以设计成最大化其大小和多样性,同时确保每个变体在测序通量限制内具有足够的读取量。
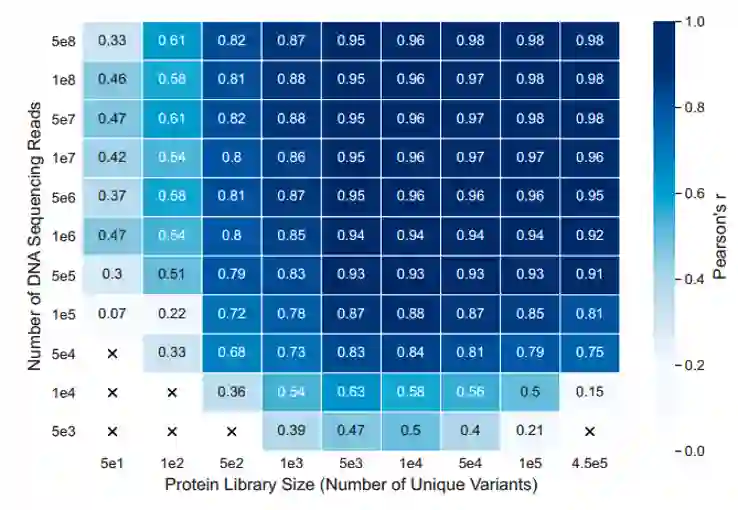
图3 文库大小和测序读取次数不同时,GB1数据集上训练的序列卷积模型的性能******[1]**********
3.模型提供了对蛋白质结构和机制的洞察
使用统一流形逼近与投影(UMAP)技术可视化GB1序列卷积网络最后一层潜在空间中的测试集序列(图4A),作者发现了具有三个突出的低分变体簇,它们具有对应破坏GB1功能的不同机制。“G1”和“G2”簇,分别包含蛋白质N和C末端附近核心残基突变的变体,可能会破坏蛋白质的结构稳定性,从而降低深度突变扫描实验中测量的活性。簇“G3”包含在IgG结合界面处具有突变的变体,可能通过破坏关键结合相互作用来降低活性。这种基于不同分子机制的变异聚类表明,网络可以学习到蛋白质功能的生物学意义。 作者用神经网络模型来检测哪些序列位置对蛋白质功能的影响最大,计算Pab1中所有训练集变体的积分梯度归因,并将这些值映射到三维结构上(图4B)。负归因表明该位置的突变降低了蛋白质的活性,关键界面残基N127具有最大的负归因,这一结果与先前报道吻合;D151具有最大的正归因,这与天冬氨酸在天然存在的Pab1序列中在位置151不常见的观察结果一致。序列卷积网络能够直接从原始数据中学习生物学相关信息,而无需指定详细的分子机制。使用Pab1序列卷积网络对所有可能的单突变变异进行预测显示了Pab1序列中对突变不耐受的区域(图4C),发现脯氨酸突变在大多数序列位置都是不利的。
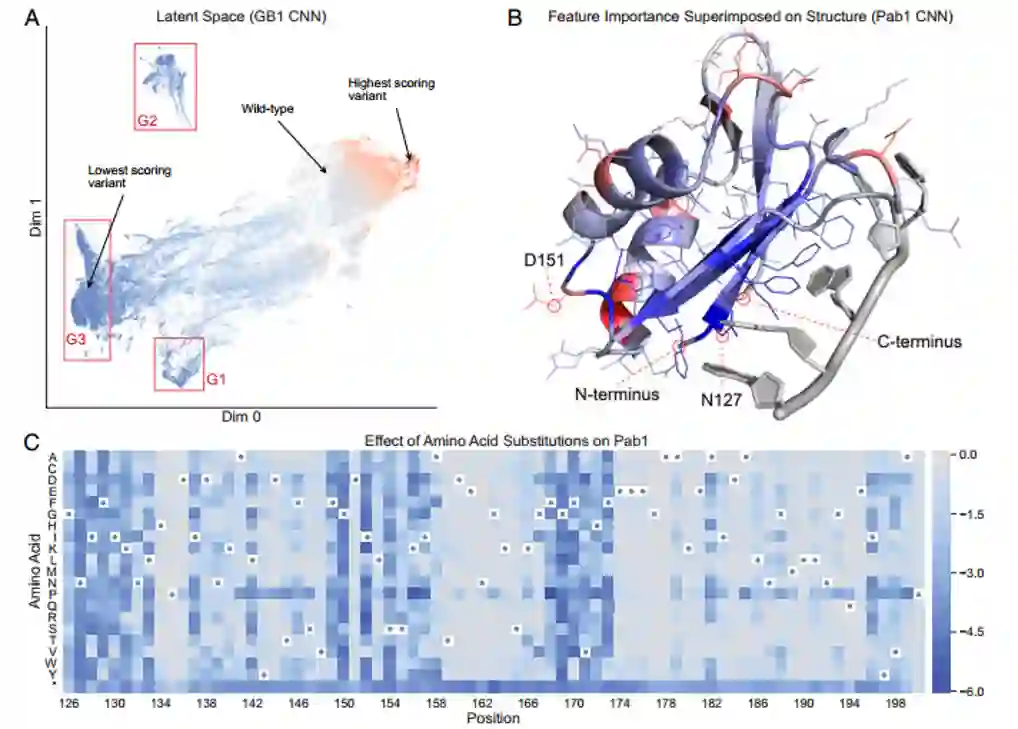
图4 (A)GB1序列卷积网络潜在空间的UMAP投影(B)积分梯度归因在Pab1结构的对应(C)所有单个突变预测的热图******[1]**********
4.使用模型设计远距离蛋白质序列
作者设计了一组与野生型序列不同的新GB1变体,用来测试监督模型的泛化能力(图5A)。使用序列优化方法设计了五个GB1变体,其中野生型突变数量(10,20,30,40,50)不断增加,代表序列一致性从82%下降到11%。对10个突变体设计,称为Design10,其为可溶性蛋白质,但更多突变数目的蛋白是不溶的。作者检测了Design10的圆二色性光谱,发现其与野生型GB1几乎相同,表明它们具有相似的二级结构含量(图5B)。Design10与IgG结合的Kd为5 nM ,其亲和力明显高于野生型GB1(图5C),与文献报道对比估计Design10结合人IgG的亲和力至少是野生型的20倍。 作者使用Rosetta de novo结构预测构建了Design10的三维结构模型(图5D)。Design10的预测结构与野生型GB1晶体结构一致,与晶体结构Cα的RMSD为0.9 Å。对Design10预测结构的检查表明,其许多突变集中在IgG结合界面附近,这可能有助于解释其IgG结合亲和力的大幅增加。
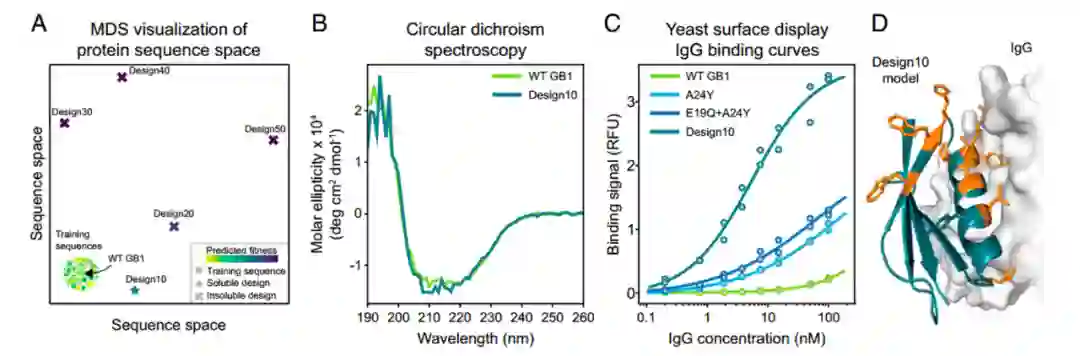
图5 基于神经网络的蛋白设计**********[1]**********小结
在本文中,作者提出的监督模型在使用大规模数据集进行训练时效果最好,当仅使用数百个序列功能样本进行训练时,模型仍优于基于物理和无监督的预测方法。不可否认的是当序列功能数据很少时无监督方法仍然很有吸引力。此外,在监督模型中,线性回归由于无法表示多个突变间的相互作用而表现最低。尽管存在局限性,线性回归仍然表现较好,原因可能是突变通常以加法方式进行组合。对数据质量如何影响学习序列功能映射能力的分析,表明模型的预测性能不仅取决于训练样本的数量,还取决于估计的功能分数的质量。因此,在深度突变扫描实验中最好限制分析的唯一变异的总数,以确保每个变异具有足够的测序读数来计算准确的功能评分。作者认为在未来可以探索如何更好地结合全局蛋白质表征,局部残基共进化特征和蛋白质结构的图形编码,从而学习特定蛋白质功能的预测模型,包括几乎没有实验数据的蛋白质。
**参考文献 **
[1] Gelman S, Fahlberg S A, Heinzelman P, et al. Neural networks to learn protein sequence–function relationships from deep mutational scanning data. Proceedings of the National Academy of Sciences, 2021, 118(48). DOI:10.1101/2020.10.25.353946 [2] Hopf T A, Ingraham J B, Poelwijk F J, et al. Mutation effects predicted from sequence co-variation. Nature biotechnology,2017, 35(2): 128-135. [3] Riesselman A J, Ingraham J B, Marks D S. Deep generative models of genetic variation capture the effects of mutations.Nature methods, 2018, 15(10): 816-822.
供稿:王保利
校稿:李诗良/沈子豪编辑:王思雨华东理工大学/上海市新药设计重点实验室/李洪林教授课题组▼招聘博后▼华东理工大学李洪林教授团队诚聘博士后