
今天给大家带来的是发表在NATURE COMMUNICATIONS上的一篇文章CopulaNet: Learning residue co-evolution directly from multiple sequence alignment for protein structure prediction。该文章提出一个端到端网络CopulaNet,直接从多序列比对(MSA)结果中推测残基协同进化,进而对残基间的距离做出更精准的预测,这对于蛋白质三级结构的预测非常重要。

1 介绍
残基协同进化已成为估计蛋白质残基间距离的主要原理,同时对预测蛋白质结构也至关重要。现有的方法大多采用间接策略,即基于一些手工计算出的特征来推断残基的协同进化,例如从MSA中计算残基间的协方差矩阵。然而,这种间接策略并不能充分利用MSA所携带的信息。因此本文提出了一个端到端的深度神经网络CopulaNet,直接利用MSA推测残基间的协同进化。 CopulaNet的关键要素包括:(i)为每个残基构建的编码器;(ii)模拟残基间协同进化的聚合器,进而估计残基之间的距离。以CASP13的目标蛋白为代表,文章证明了CopulaNet可以提高蛋白质结构预测的准确性和效率。 文章用两个人造蛋白质P1和P2作为示例(图1)来证明间接策略的一个突出缺点,即将MSA转换为手工计算的特征后会有相当大的信息损失。在蛋白质P1中,两个残基R1和R2很接近,在蛋白质P2中,它们彼此相距很远。P1和P2所构建的MSA存在差异,但从这些MSA计算出的协方差矩阵完全相同,这导致直接耦合分析(DCA)技术对P1和P2蛋白质给出了相同的距离估计。事实上,对于这两个MSA,任何一个残基的统计量,或者两个残基的成对统计量,都不能区分它们。因此,一种能够从MSA中提取残基间协同进化信息更有效的方法是非常有意义的。
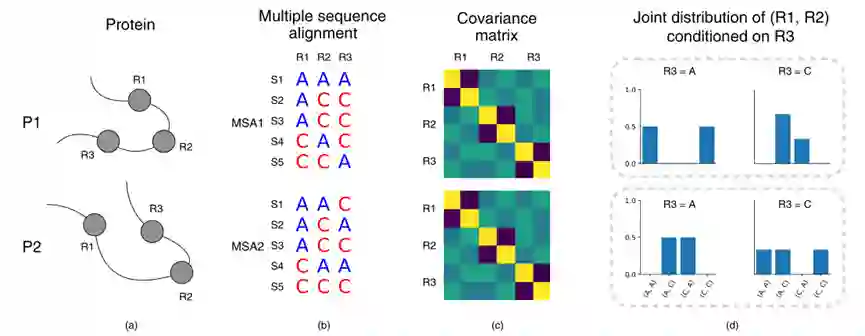
图1 基于协方差的方法在估计残基间距离时的局限性
文章提出了一个端到端的深度神经网络框架,称为CopulaNet,用于估计残基之间的距离。CopulaNet由MSA编码器、协同进化聚合器和距离估计器三个关键部件组成。MSA编码器分别处理MSA中的每个同源蛋白。对于任意两个残基,聚合器首先计算它们从每个同源蛋白中获得的嵌入特征的外积,然后使用平均池聚集从所有同源蛋白中获得的外积,最后得到两个残基之间共同进化的度量。基于得到的残基协同进化,使用2D残差网络估计残基对之间的距离。 以CopulaNet为核心模块,文章开发了一种蛋白质结构预测方法(称为ProFOLD)。简单来说,ProFOLD将估计的距离转化为势能函数,实现势能最小的三级结构构象。文章以蛋白质T0992-D1为例,展示ProFOLD的概念,然后将其应用于CASP13目标蛋白的结构预测,最后将其与目前最先进的预测方法进行比较。文章还对CopulaNet的关键元素的贡献进行了分析。
2 方法
2.1 CopulaNet架构
MSA编码器的目的是模拟每个目标蛋白残基的突变。文章将一个含有K个同源蛋白的MSA表示为K对比对,每个比对都由一个与目标蛋白对齐的同源蛋白组成。对于每个单独的比对,MSA编码器识别目标蛋白的每个残基的突变,并将突变嵌入到一个包含64个特征的向量中。 由于残基的突变与其相邻残基高度相关,MSA编码器在嵌入时应该将残基与其相邻残基一起考虑。为此,文章将编码器设计成具有多个卷积层的1D卷积残差网络,从而使其能够将一个残基与其邻居一起嵌入。 MSA编码器使用1D残差网络。残差网络有8个残差块,每个残差块由两个批量归一化层、两个具有64个滤波器的一维卷积层(核大小为3)和ELU组成。
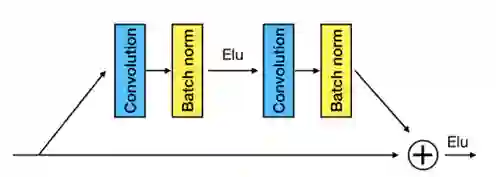
图2 MSA编码器和距离估计器使用的残差块结构
协同进化聚合器衡量两个残基之间的共同突变。 考虑一个具有L个残基t1t2⋯tL的目标蛋白,以及一个预先构建的含有K个同源蛋白的MSA。将MSA编码器应用于MSA的第k个同源蛋白上得到C×L的嵌入特征,记为Xk,其中C表示MSA编码器的输出通道数。对于目标蛋白中的残基ti,从所有同源蛋白中提取其嵌入特征聚合在一起。聚合的嵌入特征记为f,计算如下:
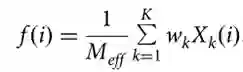
其中wk为第k个同源蛋白的权重,Meff为所有同源蛋白的权重之和。根据PSICOV惯例,权重wk计算方式为与第k个同源物具有至少80%序列相同的相似同源蛋白数量的倒数,因此Meff表示在MSA中记录的有效同源蛋白数量。 对于目标蛋白中的两个残基ti和tj,协同进化聚集器使用聚合协同进化特征h(i,j)衡量它们的共同突变。
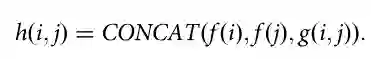
其中g(i,j)表示残基ti和tj的嵌入特征的聚合外积,计算方法如下:
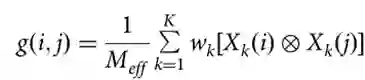
其中“⊗”表示外积操作。 综上所述,聚合协同进化特征包括C×2聚合嵌入特征和C×C聚合外积特征。本文中MSA编码器的输出通道尺寸C设置为64。因此,协同进化聚集器对目标蛋白中的任意两个残基产生总共4224个(64×2+64×64)协同进化特征。
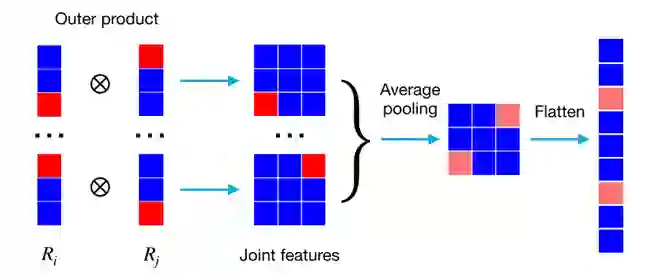
图3 在协同进化聚合器模块中嵌入特征和平均池化的外积示例
距离估计器的目的是利用72个残差块的2D残差网络,根据得到的残基协同进化来估计残基之间的距离。为了减轻距离估计的困难,文章将距离估计问题转化为分类问题。将残基距离范围划分为37个区间,即(0Å,2.5Å),(2.5Å,3.0Å),⋯,(19.5Å,20.0Å)和(20.0Å, +∞)。对于每个残基对,CopulaNet预测37个区间上的距离分布,而不是单一的估计距离值。
2.2 ProFOLD超参数设置
在综合考虑预测性能和模型规模的基础上,文章确定了ProFOLD的超参数。表1 ProFOLD超参数设置
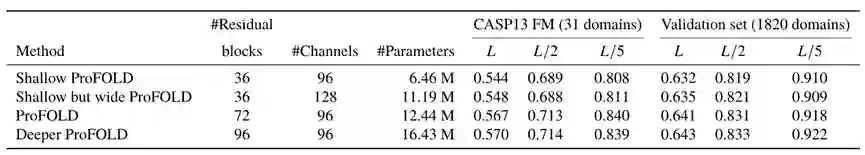
如表所示,“Shallow ProFOLD”的精度低于ProFOLD。当使用更多通道时,“Shallow but wide ProFOLD”显示出与“Shallow ProFOLD”大致相同的精度。 结果表明,与通道数相比,ProFOLD的性能对残差块数更为敏感。当进一步增加残差块的数量时,精度大致固定,但参数的数量急剧增加。 为了平衡性能和模型大小,在本文研究中使用72个残差块和96个通道的2D残差网络。
2.3 基准数据集
在研究中使用与AlphaFold相同的基准数据集。简单来说,基准数据集总共包含31247个非冗余结构域,进一步划分为训练集和验证集(分别包含29247个和1820个蛋白质)。 文章在CASP13目标上测试了方法,该目标由来自71个目标蛋白的104个域组成。这104个域分为三类:FM(31个域)、FM/TBM(12个域)和TBM(61个域)。训练集和测试集之间没有重叠。
2.4 MSA生成和表示
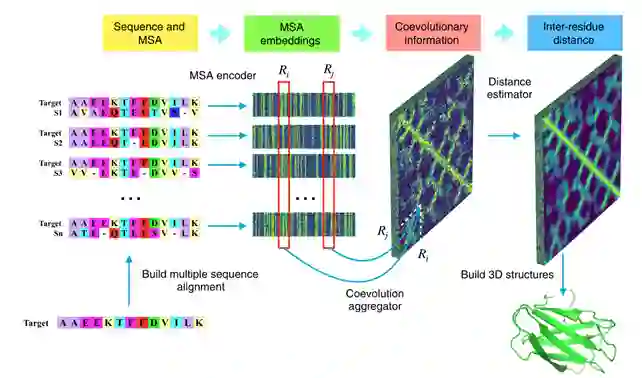
ProFOLD以目标蛋白的多序列比对作为唯一输入。在本文研究中,文章将得到的MSA表示为序列对的集合。每个序列对包含目标蛋白和一个同源蛋白。通过在比对序列中添加gap来构造两个长度相等的字符串,使匹配的字符在连续的位置对齐(图4)。然后将每个位置编码为41元素的二进制向量。
3 结果
3.1方法总结
图4总结了ProFOLD方法。以CASP13靶蛋白T0992-D1为例,文章介绍了用于蛋白质结构预测的ProFOLD的概念和主要步骤。T0992-D1蛋白共有107个残基(这里只显示了前13个残基)。对于该蛋白,首先在Uniclust30、UniRef90和Metaclust50等蛋白质序列数据库中进行了同源物的检索。得到的同源蛋白(2807个结构域)组织成一个MSA。接下来应用CopulaNet直接从构建的MSA估计残基间距离。在这里,文章推断了预定义的37个bins之间的残基距离的分布,而不是一个单一的距离值。残基LEU32和TYR70最可能的距离区间被预测为(7.5Å,8Å),它涵盖了真实距离7.83Å。最后将估计的距离分布转化为势能函数,然后通过势能函数最小化来寻找结构构象。ProFOLD报告了具有足够低的势能函数的结构构象作为最终预测结果,它完全近似于原生结构(TMscore: 0.84)。
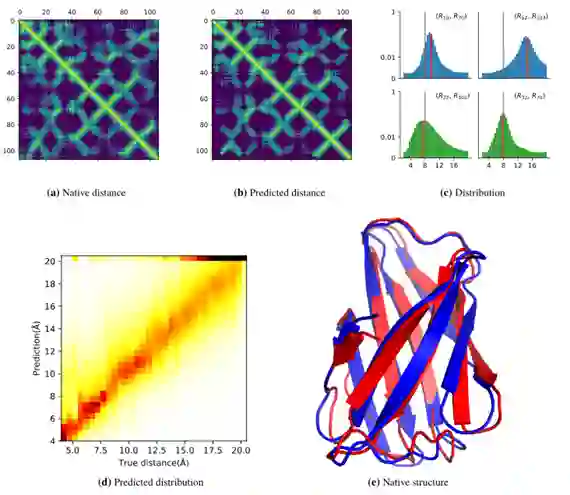
图5 利用ProFOLD预测CASP FM靶蛋白T0992-D1的三级结构
3.2 利用CopulaNet估计残基间距离
利用CopulaNet, ProFOLD估计了104个CASP13蛋白结构域的残基间距离。为了进行公平的比较,文章根据预测的残基间接触的精度而不是残基间距离来评估。具体而言,对于两个残基,将距离小于8Å的区间的预测概率相加作为两个残基接触的预测概率。如下图6所示,在31个FM域上,ProFOLD对最可能的L/5、L/2和L长程接触的预测精度分别为0.840、0.713和0.567,显著高于CASP13的优胜者组AlphaFold,分别高出0.128、0.117和0.097。文章还与更新后的RaptorX进行了比较。ProFOLD的预测精度高于现有方法。
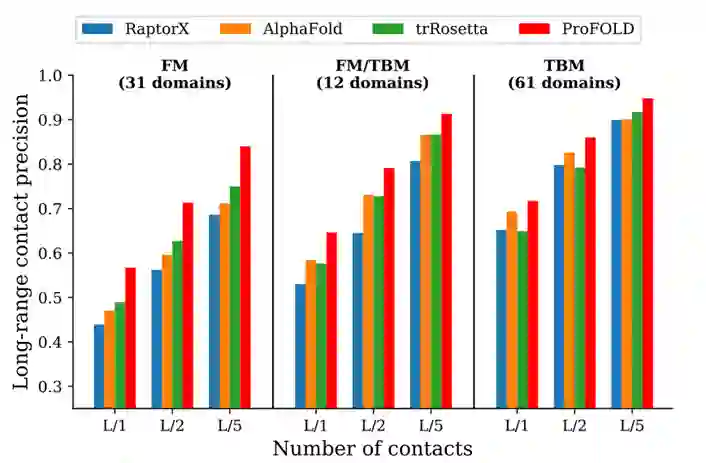
图6不同方法残基间接触精度的比较
文章进一步分析了CopulaNet的不同模块对估计残基间距离的贡献。已知CopulaNet的独特性在于使用可学习的“编码器和聚合器”框架,而不是传统的统计模型来推断残基的共同进化,得到的残基协同进化进一步输入到2D残差网络中。为了检查编码器和聚合器的贡献,通过禁用ProFOLD中的2D残差网络构建了一个包含这些组件的ProFOLD w/o R,与标准ProFOLD进行评估和比较。 文章还通过将ProFOLD的“编码器和聚合器”组件分别替换为协方差矩阵(L×L×21×21矩阵)和CCMpred输出(L×L×21×21矩阵)构造了三个模型。使用CCMpred输出的模型记为baseline-CCM,使用协方差矩阵模型记为baseline-Cov。文章进一步构造了一个模型(baseline-CF),该模型使用了综合特征,包括氨基酸类型、PSSM、预测的二级结构、互信息、协方差矩阵和CCMpred输出。 如下表中所示,在31个CASP13 FM目标上,ProFOLD在长程接触预测方面的精度高于baseline-CCM和baseline-Cov。尽管baseline-CF使用了全面的特性并显示了性能改进,ProFOLD仍然优于baseline-CF。即使使用较浅的2D残差网络,在验证集上也可以观察到ProFOLD优于这些模型。表2 ProFOLD和baseline模型在接触预测精度方面的比较
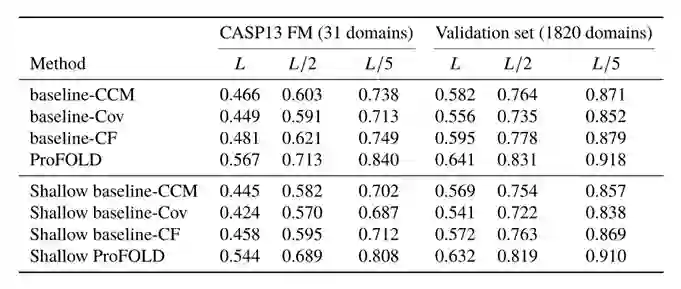
综上所述,对估计残基间距离的主要贡献来自可学习的“编码器和聚合器”框架。
3.3 利用ProFOLD预测蛋白质三级结构
文章用ProFOLD来预测蛋白质三级结构,并将其与目前最先进的方法进行比较。 如图7所示,在CASP13中的31个FM上,ProFOLD的表现优于目前最先进的方法。具体而言,当使用高质量结构的标准(TMscore≥0.70)时,ProFOLD预测了31个结构域中的18个高质量结构,而AlphaFold和trRosetta分别只预测了12个和7个高质量结构。此外,ProFOLD预测结果的平均TMscore为0.662,远高于trRosetta(0.584)和A7D(0.580)。正面比较清楚地展示了ProFOLD优于AlphaFold的优势:在31个FM域中的24个上ProFOLD优于AlphaFold。ProFOLD在27个FM目标上的表现优于trRosetta。
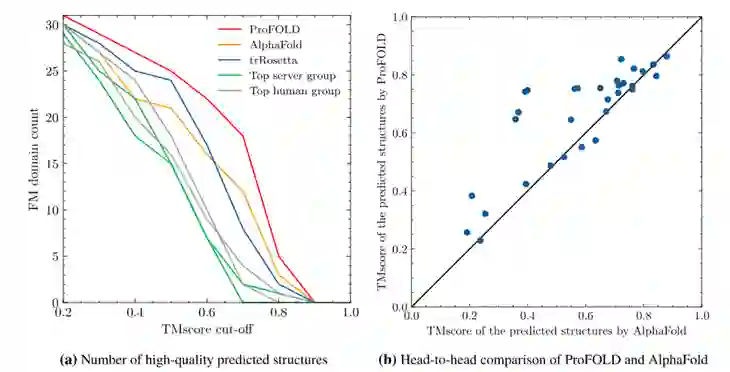
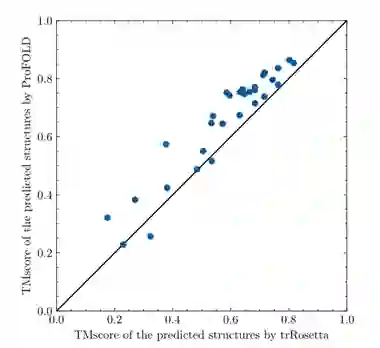
图7 CASP13 FM目标蛋白三级结构预测的质量
文章进一步探讨了可能影响ProFOLD成功应用的因素。先前的研究已经表明,预测结构的质量与MSA中有效同源蛋白Meff数量高度相关。如图8a所示,Meff的对数与ProFOLD预测结构质量的相关系数高达0.69。因此,只要目标蛋白的Meff大于20,该蛋白预测结构的TMscore就有望大于0.60,且具有较高的置信度。 对于一种蛋白质结构预测方法,一个重要的问题是能否提前判断其预测结果的质量。当已知目标蛋白的天然结构时,可以通过将其与天然结构进行比较来很容易地评估预测的结构;然而,当天然结构不可用时,事情将变得具有挑战性。文章中对于ProFOLD预测的每个结构计算top L预测接触的平均概率(记为PPC),并将其作为预测结构的质量估计。如图8b所示,预测结构的PPC与TMscore的相关性为0.82。这种强相关性能够提前判断ProFOLD预测结构的质量。具体来说,如果目标蛋白的PPC估计超过0.60,那么ProFOLD预测结构的TMscore预计将超过0.60,且具有较高的置信度。
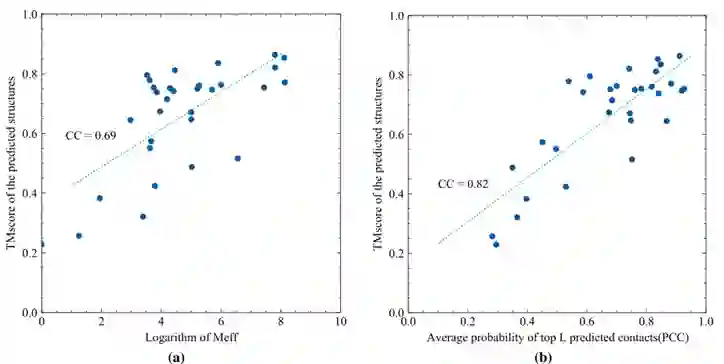
图8 影响ProFold性能的因素
4 讨论
使用ProFOLD进行蛋白质结构预测的结果突出了直接从MSA学习残基共同进化的特点。使用CASP13目标蛋白作为代表,文章所提方法的能力已经得到了明确的证明,预测结构的质量有所提高。使用端到端CopulaNet框架,ProFOLD可以准确估计残基之间的距离,从而预测蛋白质结构。ProFOLD的效率提高是另一个优势,主要是由于CopulaNet的简洁架构。需要指出的是,CopulaNet的基本思想和架构可以很容易地修改,以计算除残基协同进化之外的其他领域的条件联合分布。 综上所述,直接从MSA中学习残基协同进化的工作,以及最近在构建高质量MSA方面的进展,将有助于更准确地预测蛋白质三级结构,进而理解蛋白质功能。
