本文介绍一篇浙江大学药学院教授及碳硅智慧首席科学家侯廷军教授团队最新发表在Journal of Medicinal Chemistry期刊上的研究成果—— “Boosting Protein-Ligand Binding Pose Prediction and Virtual Screening Based on Residue-Atom Distance Likelihood Potential and Graph Transformer”。本文提出了一种新型蛋白-配体结合强度的打分函数RTMScore,该打分方法通过Graph Transformer提取蛋白质的氨基酸残基以及配体的原子节点特征,并通过混合密度网络(Mixture Density Network, MDN)获取蛋白质各氨基酸残基和配体各原子间距离的概率密度分布,最后将其转化为统计势以用于蛋白-配体间结合强度的评估。RTMScore的对接和筛选能力在CASF-2016标准数据集上显著超越了当前的其他主流方法,同时能够作为一种有效的重打分工具用于大规模虚拟筛选。本文代码和模型已经开源。

1 研究背景 一直以来,如何准确评估蛋白质与配体之间的结合模式及结合强度都是基于结构的药物设计领域的核心问题。相比于成本高但效率低的实验方法,分子对接技术可以快速预测靶标与配体之间的结合模式并评估其结合强度,因此已成为先导药物分子发现的核心技术。传统上,分子对接技术,首先基于构象搜索算法对配体分子的构象进行采样,然后采用打分函数(Scoring Function, SF)来评估蛋白与配体分子各个构象的结合强度,通常得分最高的构象被认为是最合理的配体分子的结合构象。近年来,分子对接技术在药物设计和发现中发挥了重要的作用,但是也存在着很大的局限性,大部分打分函数的预测精度有限,不能很好地满足虚拟筛选的需求。
随着机器学习(Machine Learning, ML)技术的不断发展,大批基于机器学习的打分函数(Machine Learning-based SF,MLSF)被报道。这些打分函数显示出了比传统方法显著更优的性能,但大部分方法依然存在着泛化能力差、应用范围窄等问题。因此开发出预测精度更高且泛化能力更强的打分函数对于提高药物分子设计的效率极为重要。
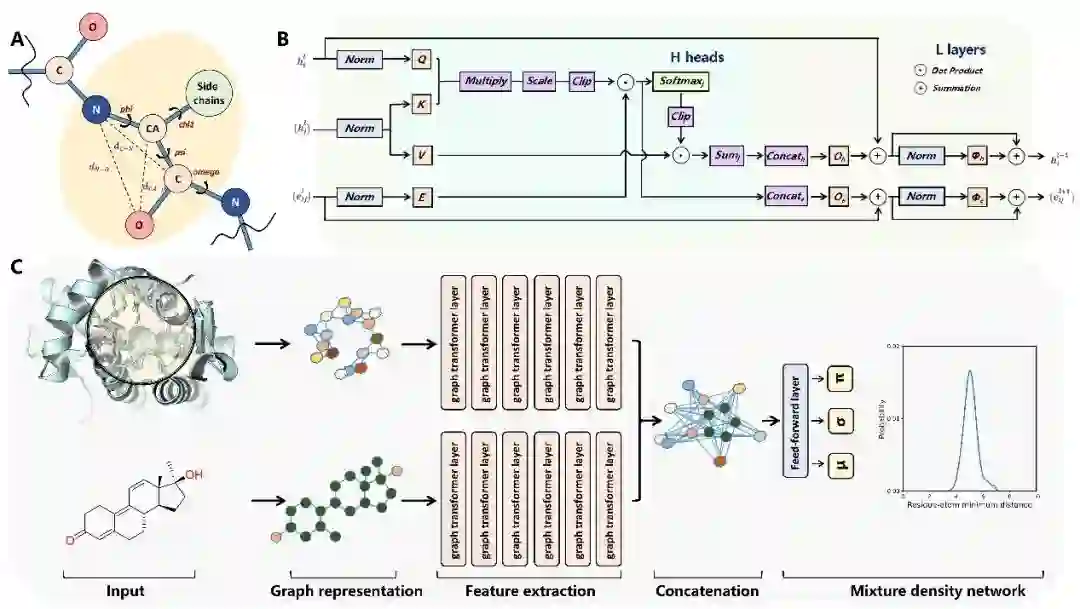
2 方法 RTMScore的构建流程如图1所示,它主要包含三个模块:特征提取模块、特征拼接模块和混合密度网络。在特征提取模块,蛋白质的口袋和配体小分子被分别表示为三维残基图和二维分子图,随后通过堆叠多层的Graph Transformer分别提取口袋中的氨基酸残基和配体分子中各原子的节点特征。在特征拼接模块,蛋白质口袋和配体分子的节点特征被两两拼接起来,输入到混合密度网络中以计算出拟合混合密度模型所需的参数。通过该模型可获取蛋白质口袋中各氨基酸残基与配体分子中各原子的最短距离概率分布,再将所有统计值以负对数似然的形式进行加和,即可得到用于表示最终蛋白-配体结合能力的统计势。
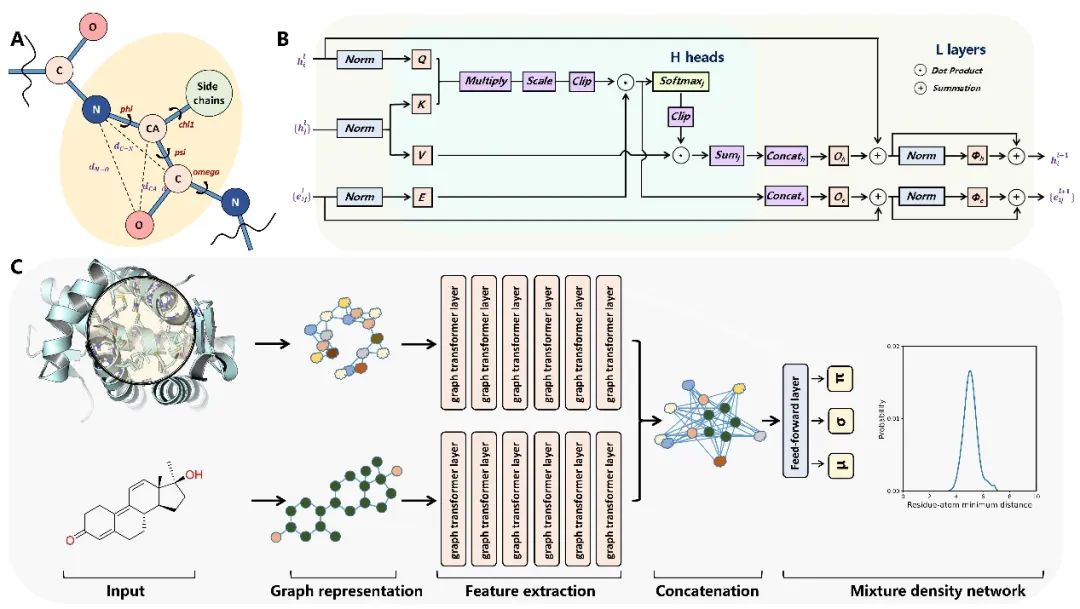
图1. RTMScore的构建流程示意图
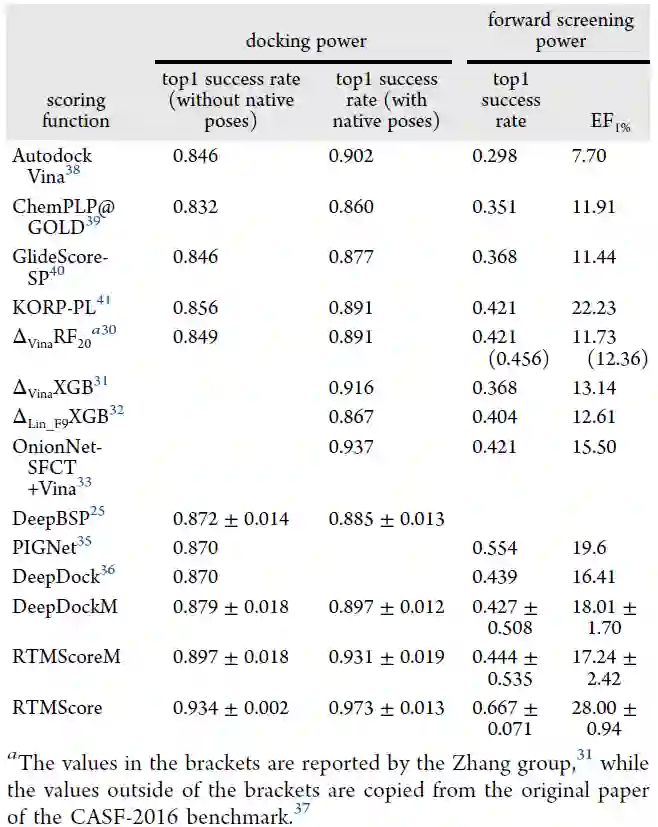
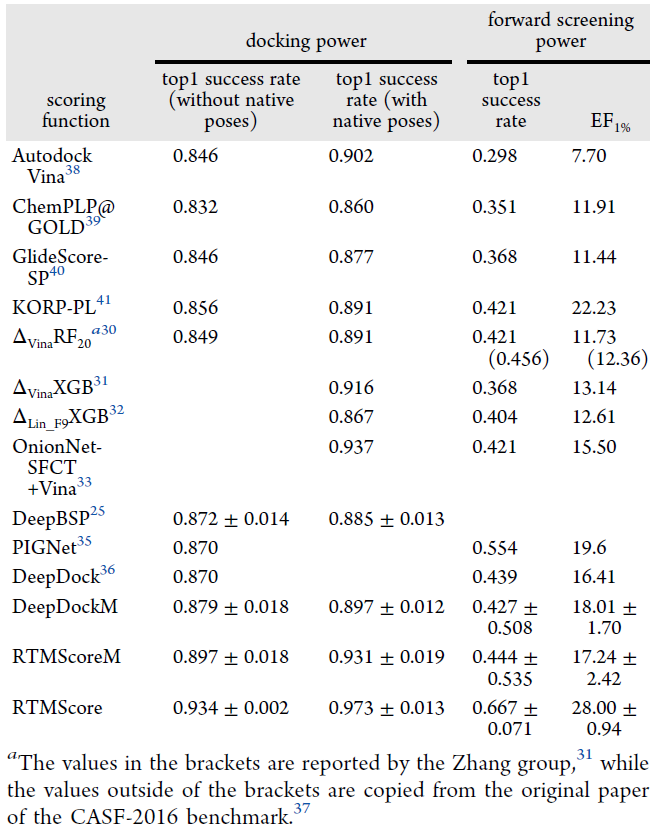
3 实验结果 CASF-2016数据集 作者在打分函数评价的基准数据集CASF-2016上评估了RTMScore的对接和筛选能力,并与多种主流方法进行了对比。如表1所示,RTMScore的平均top1对接成功率可在有无天然构象存在的条件下下分别达到97.3%和93.4%(PIGNet和DeepDock在无天然构象时仅为87.0%);另一方面,在筛选能力评估中,RTMScore的平均top1成功率为66.7%,1%富集因子为28.00(PIGNet和DeepDock的两项指标分别为55.4%、19.60和43.9%、16.41)。
表1. CASF-2016上的对接和筛选能力评估
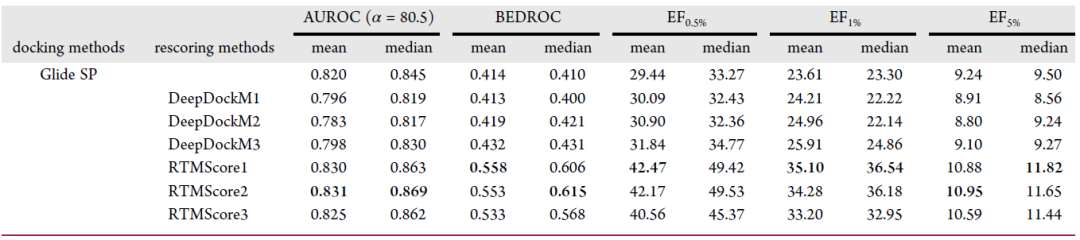
交叉对接构象数据集 考虑到CASF-2016中的对接构象均为较为理想的重对接构象,难以反映出真实场景下蛋白柔性对于对接的影响,作者接下来用Surflex-Dock、Glide SP或AutoDock Vina构造了对接构象,从而构建了交叉对接数据集PDBbind-CrossDocked-Core,在该数据集上进一步评估了RTMScore的对接能力。如表2所示,RTMScore在大部分情况下超越了传统方法和基于XGBoost构建的构象分类器,尤其是对于交叉对接构象。
表2. PDBbind-CrossDocked-Core上的对接能力评估
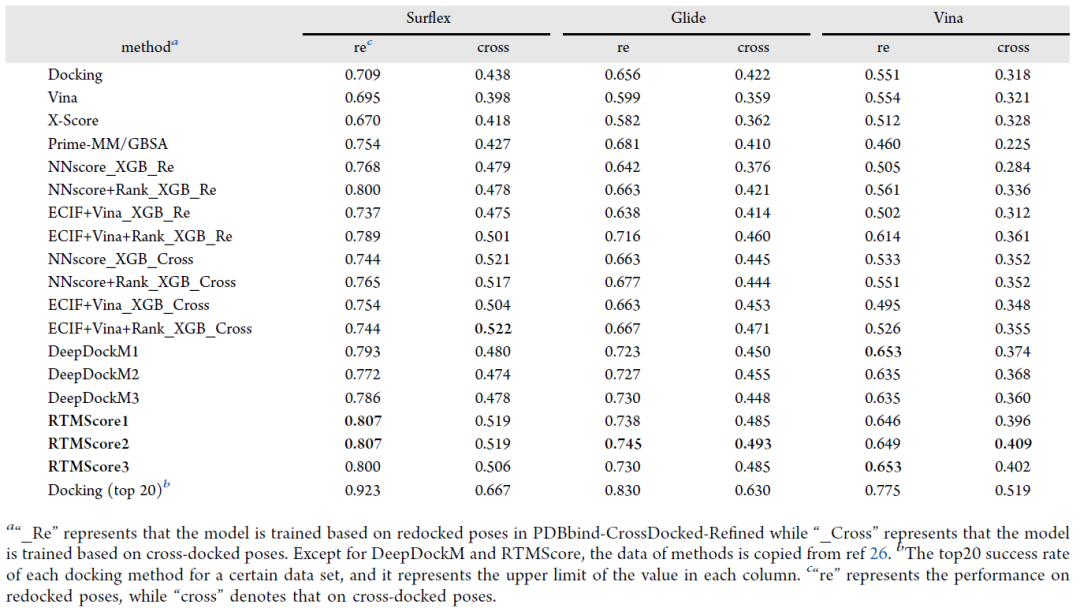
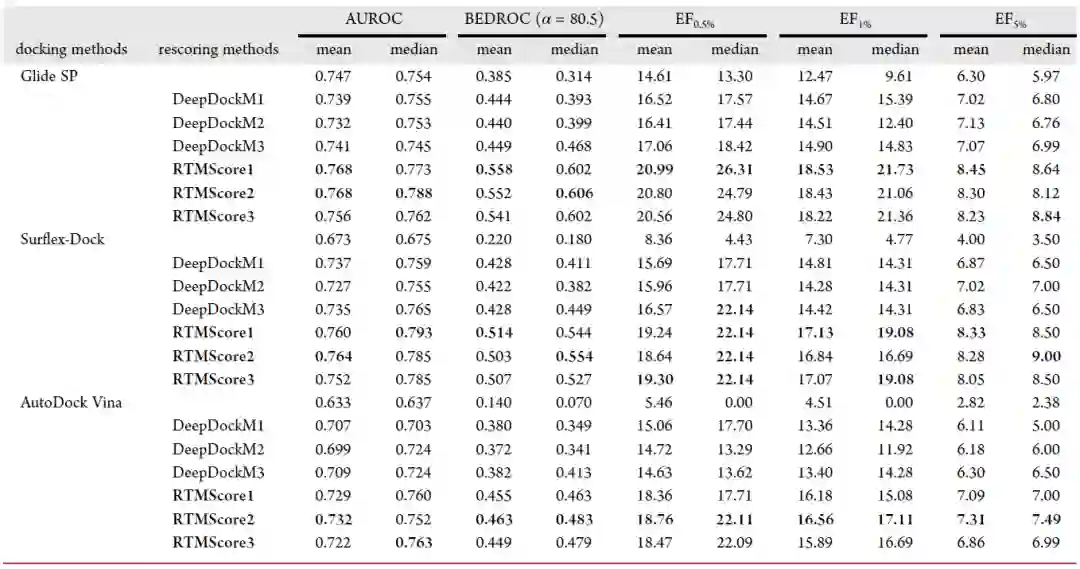
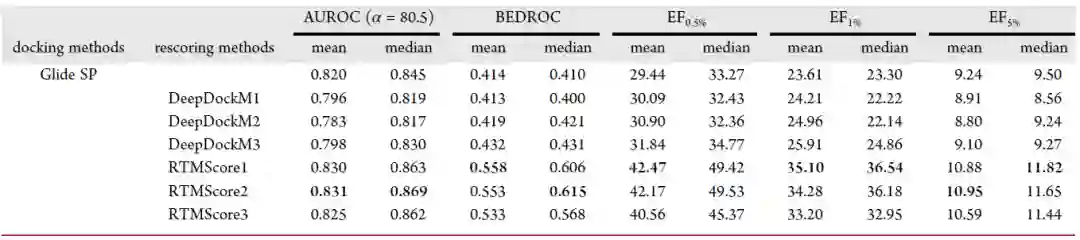
虚拟筛选数据集 接下来,作者在两个虚拟筛选数据集DEKOIS2.0(表3)和DUD-E(表4)上进一步测试了RTMScore的筛选能力。结果表明,其富集能力明显优于以相似策略构建所得的DeepDock和经典方法Glide SP。
表3. DEKOIS2.0上的筛选能力评估
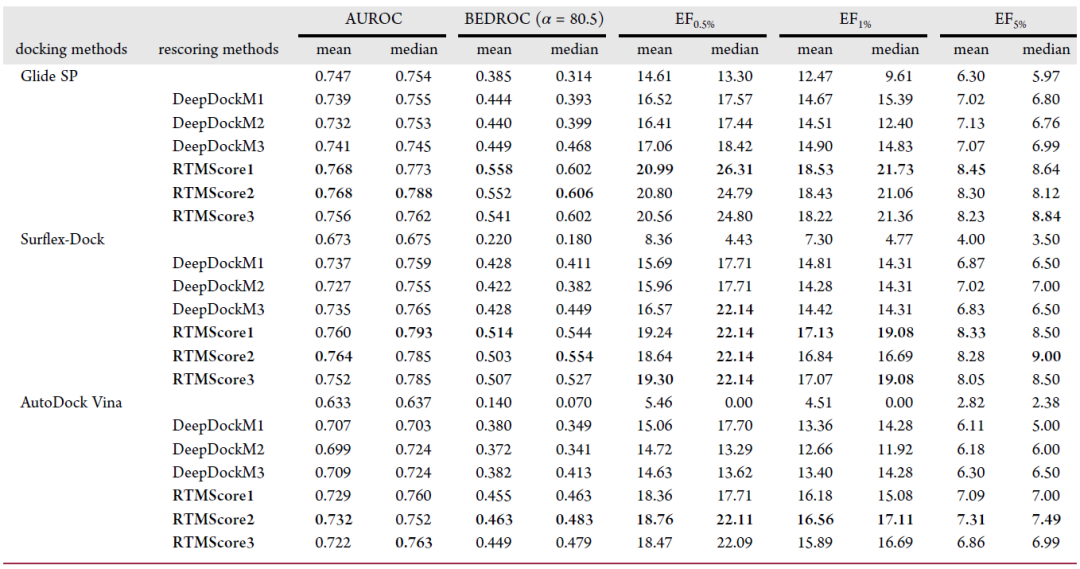
表4. DUD-E上的筛选能力评估
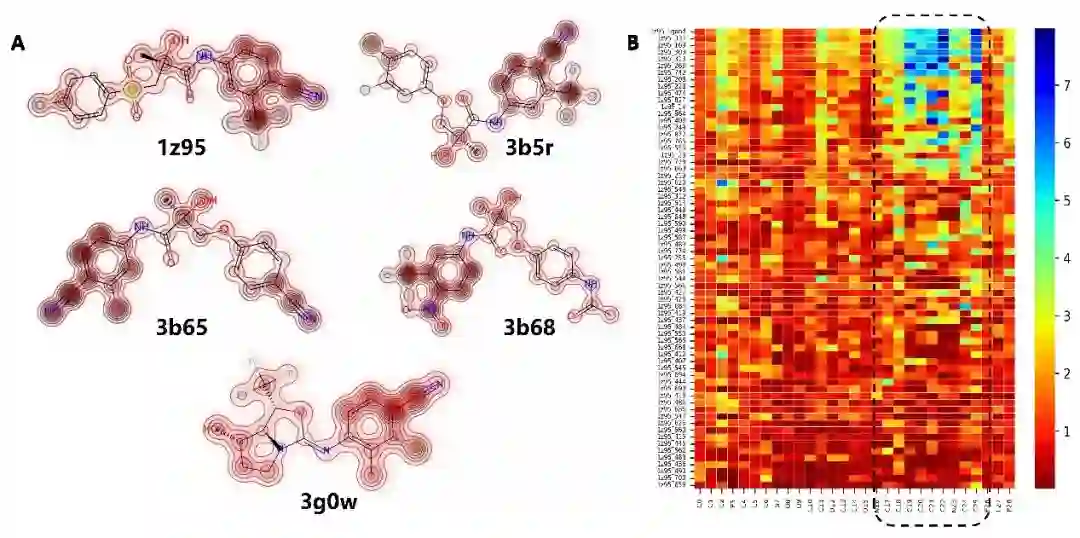
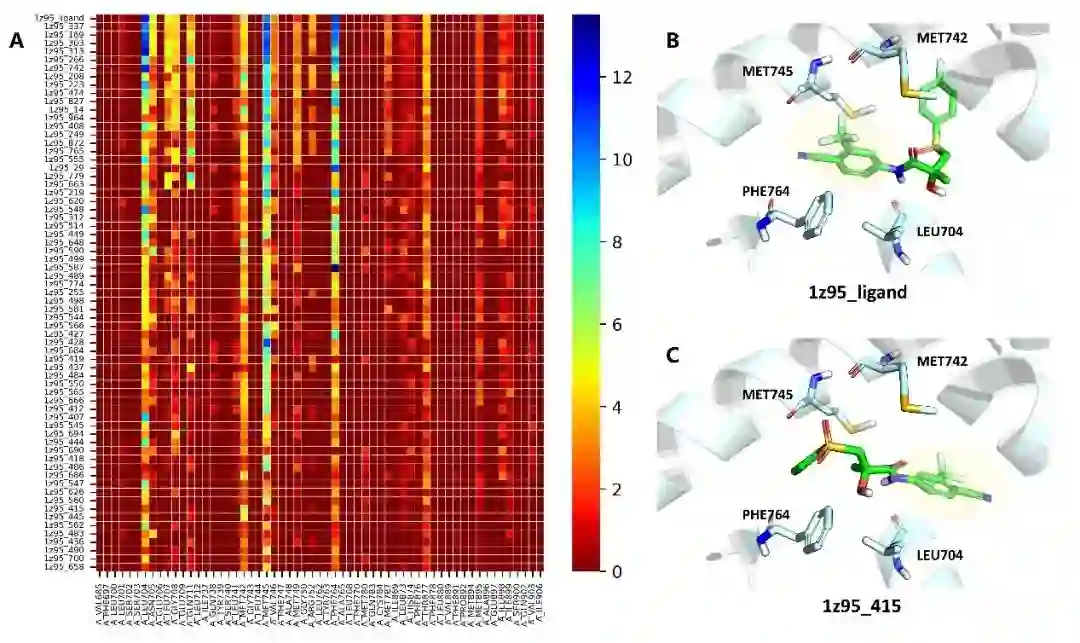
可解释性 由于RTMScore以加和的形式将各个独立的残基-原子距离似然值整合成最终的统计势,它的得分可以分别从配体原子层面(图2)或蛋白残基层面(图3)进行分解,因此RTMScore具有一定的可解释性。
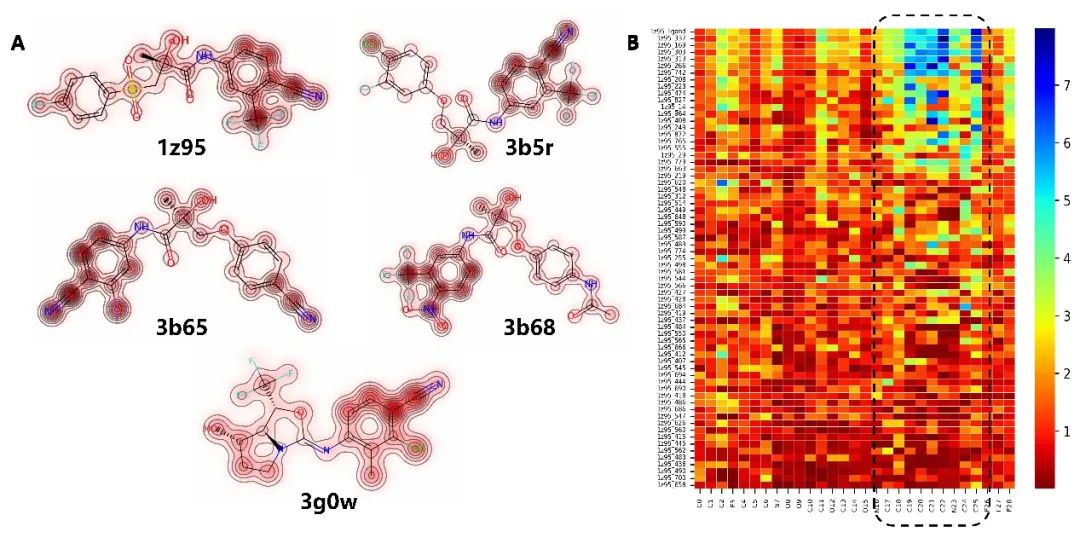
图2. RTMScore可在配体原子层面对得分进行分解
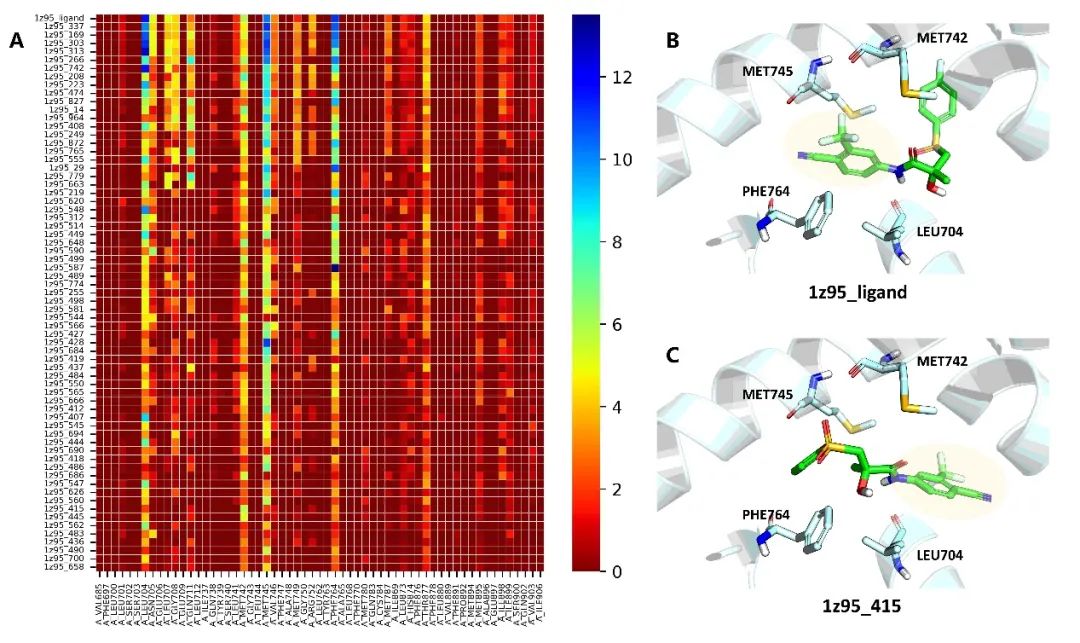
图3. RTMScore可在蛋白残基层面对得分进行分解
4 总结 作者提出了一个新型打分函数RTMScore,该方法在对接和筛选任务的预测精度和泛化能力均显著优于传统方法,有望成为基于结构的药物设计中的重要工具。此外,RTMScore可以与其他分子对接程序结合,用于结合构象预测或虚拟筛选中的重打分,也可以与一些已有对接工具进行整合,以构建新型分子对接程序。
代码和模型已经开源,请参考: https://github.com/sc8668/RTMScore。
关于DrugFlow (www.drugflow.com) DrugFlow是一个AI驱动的新药发现SaaS平台,目前供科研工作者免费试用,其创造性地将人工智能与物理计算技术深度结合,在提升底层计算模型精度的同时,还提供了优秀的数据管理能力,以此构建了一个涵盖靶标发现与验证,先导化合物发现和先导化合物优化等药物发现全过程的一站式计算平台。本文中提到的方法RTMScore未来也会上线到平台上供大家使用。 参考资料 Chao Shen, Xujun Zhang, Yafeng Deng, Junbo Gao, Dong Wang, Lei Xu, Peichen Pan, Tingjun Hou, Yu Kang, Boosting Protein−Ligand Binding Pose Prediction and Virtual Screening Based on Residue−Atom Distance Likelihood Potential and Graph Transformer, J Med Chem, 2022, DOI: 10.1021/acs.jmedchem.2c00991.