作者 | 杨崇周 指导 | 闵小平(厦门大学)
今天为大家带来的是华盛顿大学大卫·贝克实验室发表在nature communications上的文章《Sampling of structure and sequence space of small protein folds》。作者团队开发并实验验证了一个计算平台,该平台可以在采样形状多样性的同时设计多种小蛋白质折叠;设计并评估了8个不同折叠的约30000个从头蛋白质设计的稳定性。在这些设计中,鉴定了约6200种稳定的蛋白质,其中包括一些预测具有同类最小化硫氧还蛋白折叠的蛋白质;作者团队利用这个数据集开发了一种精确的分类器来预测我们设计的蛋白质的稳定性。

介绍
自然序列空间只涵盖了一小部分可能的蛋白质。分子功能的进化通常是通过相对少量已知蛋白质家族的多样化来实现的。这突出了形状多样性增加功能多样性的能力。因此,以高精度采样和控制微小结构变化的能力代表了设计具有新功能的蛋白质的重要进步。到目前为止,已经产生了一些新设计的球状折叠。然而,由于缺乏通用的计算基础设施,给定褶皱内的结构多样性还没有被特意采样和实验验证。最近的进展表明,探索环形螺旋-环形图案能够对现有折叠进行几何采样;在这里,作者团队超越并描述了如何在大范围内耗尽小蛋白质折叠的可塑性,对每个二级结构和环连接器进行采样,以在给定折叠内产生形状多样性。
随着寡核苷酸合成和实验筛选技术的出现,数千个短基因可以在大库中以单一寡核苷酸的形式生产,并进行稳定性筛选。还可以通过组装方法合成多达110个氨基酸的蛋白质。
在这里,作者团队为从头开始大规模并行设计蛋白质提供了计算管道,以快速探索蛋白质折叠的形状多样性,并利用高通量实验方法对其进行评估。与在折叠轨迹期间仅对构象进行采样的其他方法不同,作者团队的方法也在每个轨迹期间对拓扑进行采样,而无需事先了解每个残基的残基特征。它能够设计给定折叠的不同表示,并允许对(1)每个次级结构元件的长度进行采样;(2) 二级结构元件之间的距离(也可以作为分布进行评估);(3) α-螺旋和β-链的排列,如果涉及两条以上的链,则其寄存器移位;最后(4)引入和放置凸起,以将曲率引入β-绞合线。该框架自动将先前发现的蛋白质折叠规则和序列偏差应用于环连接,并可扩展到新的结构特征.
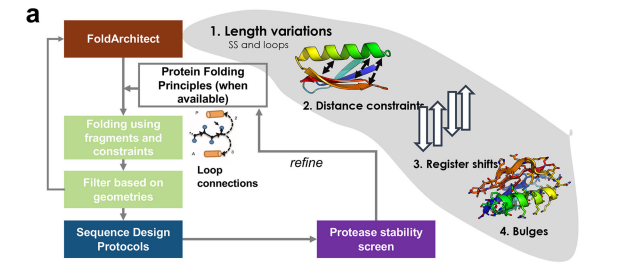
蛋白设计流程
设计结果
设计蛋白质的高通量评估 作者团队使用酵母表面展示对31500个反映这八个折叠的序列进行了实验表征,包括约2300个随机序列作为阴性对照。为了估计蛋白质稳定性,对酵母上显示的所有设计蛋白质的库进行胰蛋白酶和糜蛋白酶的滴定,并将未分离的蛋白质分类到每个蛋白酶浓度的池中。先前建立了消化蛋白质变体的EC50值与其自由能之间的直接关系。
使用荧光活化细胞分选(方法)进行选择;作者团队使用下一代测序对每个池中的序列进行计数。与之前发布的方法相比,还进行了额外的选择和重复,以提高准确性。使用通过测序获得的计数,作者团队拟合了两种蛋白酶超过31180个序列(包括对照序列)的EC50值,并计算了每个蛋白酶的稳定性得分。为了提高测定之间的可比性,作者团队使用与“稳定性分数阶梯”内部控制相同的基于蛋白酶的稳定性评估程序,添加了五种蛋白质,这些蛋白质跨越了先前测量的广泛稳定性分数。该阶梯允许调整每批酶的活性。
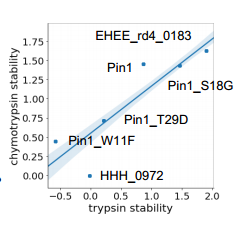
蛋白稳定性评估评估得分
设计折叠家族的评估 使用作者团队的蛋白酶消化试验,不同折叠的成功率不同(稳定性评分>0.5的设计分数)。三个螺旋束通常显示出最高的成功率;这与之前的报告一致。发明的褶皱F2和F4的成功率高于包含α螺旋和β链的自然褶皱;硫氧还蛋白和超螺旋卷曲的四个螺旋束(线圈)最低。
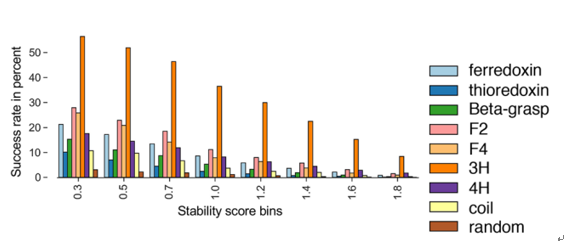
不同折叠的成功率
为了评估形状多样性,我们比较了蛋白酶稳定蛋白和不稳定蛋白的每个折叠。我们比较了二级结构元件之间的距离、寄存器移位、外β链的二面体,以描述片材的曲率、相邻二级结构元素之间的二面面、螺旋间距和螺旋角,给出了其环中的特定phi和psi角,证明了每个褶皱都具有局部塑性,验证了我们从头设计蛋白质的基本规则,并提出了一套额外的规则。
硫氧还蛋白折叠的从头设计 迄今为止,尚未报道硫氧还蛋白折叠的从头设计。
作者团队解决了更具挑战性的问题,即最小化总体折叠。天然存在的硫氧还蛋白具有三层β/α夹层,中心层由五股线形成,每侧有两个α-螺旋。许多硫氧还蛋白样蛋白在其α-螺旋或第五β链中存在变化。硫氧还蛋白折叠的保守核心元素可以细分为N端βαβ基序和C端ββα基序,它们通常由一个小螺旋连接。βαβ元素(α/β家族的特征)存在于许多较大的蛋白质中,因为它是连接基序,能够扩展蛋白质结构域空间,并将该超家族的成员与没有该基序重复的α+β家族区分开来。它的结合通常允许折叠的扩展,从而提供构建更大蛋白质的方法。因此,设计这种具有高度形状多样性的元件的能力为构建更大、形状更多样的折叠和域提供了工具。作者团队设计了硫氧还蛋白折叠的最小版本,仅包含核心四个薄片和两个平行或反平行螺旋,用扩展的环取代了常见的α2螺旋。
我们使用核磁共振(NMR)解决了我们的一个设计的结构。NMR系综与设计的模型一致,与模型相比,顶部结构的RMSD为1.9Å(基于C-α原子);偏差主要来自最后一个螺旋。所有β链都接近设计模型(1.1ÅC-alpha RMSD),这是第一个新设计的硫氧还蛋白折叠。
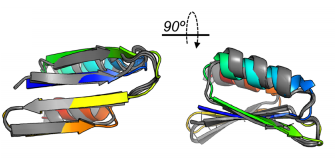
NMR结构(灰色)系综(PDB 7LDF)与Thio_802的模型(从N端(蓝色)到C端(红色)
识别稳定设计的单机学习模型 有了一组具有不同形状和物理性质的不同折叠的稳定和不稳定蛋白质,作者团队能够重新评估稳定性定义特征,并基于随机森林模型开发分类器,以确定给定的小蛋白质是否以高精度稳定。与先前描述的稳定性的物理和统计特征相比,作者团队评估了描述残基相互作用网络的额外特征以及紧密连接的残基中枢内单个氨基酸的能量贡献,总共产生了110个基于序列和结构的特征。最能预测这些附加特征的是最相关残基的总能量贡献:接触许多其他残基的残基是相互作用的枢纽。它们通常被高度掩埋,并提供蛋白质疏水核心的“粘合剂”。因此,这些“中枢”残基的有利能量学对于蛋白质核心的形成至关重要,而潜在的冲突可能导致不稳定。如前所述,通过与短结构片段对齐测量的正确局部几何结构是新设计蛋白质折叠的最具预测性特征,其次是蛋白质核心内疏水残基的数量。与之前的研究不同,通过使用更大、更多样的折叠数据集和描述性特征,作者团队能够在多个折叠上训练模型,而不是一次一个折叠,甚至可以预测不可见折叠的稳定性。作者相信,这种多样的支架集合使他们能够了解这些折叠的更一般的描述,并提高模型的预测能力。为了证实这一观察,他们预测了Rocklin等人先前发表的具有几个不同小褶皱的数据集的稳定性。并使用他们的单一分类器观察到AUC的预测能力约为0.83。
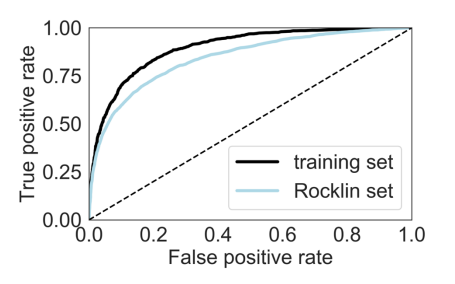
稳定性预测AUC曲线
讨论
尽管之前的工作使用基于蓝图的精心设计方案,结合人工定义的多步骤组装更大的折叠,模拟了少数折叠3–5的空间,但只有少数设计在解决方案中得到了实验验证,除了在基于蛋白酶的高通量筛开发过程中检查的小蛋白。作者团队建立在先前发现的蛋白质设计规则(包括链和螺旋的连接规则)的基础上,并提供了一种多功能折叠组件和设计管道,允许在硅折叠轨迹期间对给定折叠进行动态采样。作者团队的广泛抽样和高通量评估使他们能够同时检查数千种设计,揭示出不同的几何多样性。他们还提取了蛋白质设计的附加规则,例如螺旋元件的回路连接器,将其结合到设计算法中,并且可以在任何Rosetta设计步骤中轻松访问。最后,作者团队的广泛研究使他们能够开发一个简单的预测模型,以帮助未来的设计方法识别稳定的蛋白。
方法
三个步骤设计蛋白质。首先,构造了主干,这构成了最终折叠的三维结构。这一步骤广泛使用了这里开发的管道,与以前使用蓝图生成器的方法不同。第二步涉及序列设计,作者团队使用了两种不同的协议。第三步是选择要测试的设计。
骨架设计 Fold Architect(FoldArchitectMover)的底层算法具有多个模块化组件,它们共同设计折叠的从头肽骨架。这些组成部分共同提供了一个框架,以进行折叠级描述(例如,3个螺旋束,每个螺旋长度为10-15 a.a.,由3-4 a.a.的环连接),并产生具有所需二级结构、真实几何结构和螺旋膨胀相互作用的蛋白质骨架。
Loop环采样 没有对给定环长度的所有可能环构象进行采样,而是仅对先前发现的αβ、βα和ββ连接之间常见的环构象进行了采样。此外,作者团队的研究确定了螺旋元件连接的规则,我们也将其纳入其中。随着更多原则的确定,可以很容易地添加这些原则。此外,作者团队还提供了在相邻元素之间提供距离约束的可能性,并将“凸起”引入到股线中,他们利用这一点来构建beta抓取。先前发现的回路连接的蛋白质设计原则得到了尊重,蛋白质是动态组装的,每次添加一个片段。 二级结构元素配对 在所需折叠中相互作用的二级结构元素通过“配对”来识别。移动器和过滤器也可以使用这些信息来获得有关所需折叠的信息。不同的配对类型是“螺旋对”,它描述了两个螺旋之间的配对(例如,平行/反平行);以及“StrandPairing”,它描述了strandstrand配对(例如,并行/反并行、寄存器移位);以及“螺旋片配对”,它描述了螺旋和β片之间的相互作用。
距离约束 在不知道动态算法中使用的最终长度的情况下,可以模糊地应用任何类型的距离约束。作者团队开发了一个移动器,允许在残差选择器之间应用距离约束(DistanceConstraintGenerator)。因为褶皱是动态构建的,所以在组装过程之前不知道关键特征,例如每个二级结构元件的尺寸和N或C末端残基。
为了解决这个问题,作者团队开发了一个NamedSegment残差选择器,它跟踪每个元素,并根据需要跟踪残差编号。
即使不精确知道褶皱的最终结构组成,这也允许指定距离约束。距离约束被耦合到具有2Å的松散标准偏差的简单谐波或有界谐波函数。在测量了几个示例β-抓取和铁氧还蛋白折叠的螺旋和薄片之间的距离后,作者团队选择了8Å作为薄片和螺旋距离的默认起始值。根据给定折叠的输出进行调整,并增加或减少距离。
序列设计 代表完整折叠的骨干结构在设计其序列之前严格过滤欧米伽角和拉玛角。使用了两种不同的设计方案,一种利用先前描述的成对运动来设计蛋白质的核心。配对基序数据库包含从晶体结构中提取的两个氨基酸的两个直接相互作用的侧链,因此描述了一个“配对”。作者团队观察到,使用该协议,观察到高效的序列设计通过了所有过滤器,如局部结构几何、确保良好包装的特定半径内的平均连接度、Rosetta分数等。
参考资料 Linsky, T.W., Noble, K., Tobin, A.R. et al. Sampling of structure and sequence space of small protein folds. Nat Commun 13, 7151 (2022). https://doi.org/10.1038/s41467-022-34937-8
项目 https://github.com/strauchlab/scaffold_design