今天给大家带来的是发表在PLOS COMPUTATIONAL BIOLOGY上的一篇文章Fast and accurate Ab Initio Protein structure prediction using deep learning potentials。尽管近年来在蛋白质结构预测方面取得了巨大的进展,但对于缺乏同源序列或结构的蛋白质建模精度仍有待提高。文章提出了DeepFold,该方法集成了深度残差神经网络预测的空间约束以及基于知识的能量函数,以指导其梯度下降折叠过程。结果表明,DeepFold创建的模型准确性大大超过了经典折叠方法和其他领先的深度学习方法。尤其是在同源序列很少的目标上的建模性能方面,DeepFold的TM平均得分比trRosetta高40.3%,比DMPfold高44.9%。此外,DeepFold比传统的碎片组装快262倍。

**1.**介绍
蛋白质结构预测的目标是根据蛋白质序列确定蛋白质中每个原子的空间位置。根据PDB中是否有可靠的模板,蛋白质结构预测方法分为基于模板建模(TBM)和无模板建模(FM)。多年来,TBM一直是蛋白质结构建模最可靠的方法。然而,它的准确性本质上取决于同源模板的可用性和目标-模板比对的质量。从头建模使用先进的能量函数和采样技术来提高PDB中缺乏同源模板时蛋白质的折叠性能。在过去的几年里,使用深度学习技术从序列或多序列比对(MSA)中预测空间约束极大地提高了从头预测的准确性。CASP14将多个约束,包括距离图、残基二面角和氢键与折叠模拟集成在一起。结果表明,与基于接触的碎片组装方法相比,引入更精确的空间信息来指导折叠方法有显著的改进。尽管建模精度得到了提高,但建立在传统碎片组装折叠技术上的方法,如I-TASSER、Rosetta和QUARK,通常需要很长的模拟时间,特别是对于较长的蛋白质。事实上,从头建模需要大量的构象采样,这是由于与蛋白质折叠相关的巨大结构空间和复杂的能量景观。先进的深度学习技术可以提供丰富的高质量约束,这些准确的约束可以在很大程度上平滑粗糙的蛋白质折叠能量景观。受这些启发,文章开发了一个快速的蛋白质折叠方法--DeepFold,它结合了基于知识的统计力场和由DeepPotential产生的深度势能,目的是提高蛋白质结构从头预测的速度和准确性。该方法在大规模数据集上进行了基准测试,显示出了优于其他领先预测方法的优势,与传统的折叠模拟方法相比大大减少了模拟时间。
**2.**方法
DeepFold是一种利用深度学习约束快速构建精确全长蛋白质结构模型的算法,包括三个主要步骤:由DeepMSA2生成MSA、由DeepPotential预测空间约束以及进行L-BFGS折叠模拟,如下图所示。
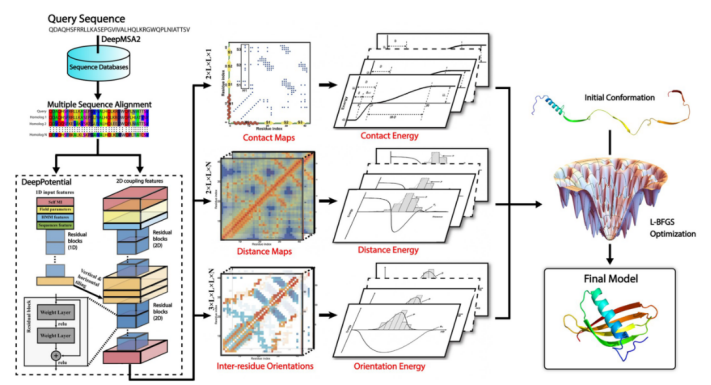
图1.DeepFold概述
由DeepMSA2生成MSA:
如下图所示,DeepMSA2通过迭代搜索全基因组(Uniclust30和UniRef90)和宏基因组(Metaclust, BFD和Mgnify)序列数据库来收集7个候选MSA。在dMSA阶段,目标序列首先由HHblits2通过Uniclust30(2017_04)进行搜索,以创建MSA-1。接下来,使用Jackhmmer和HMMsearch识别的序列构建一个自定义的HHblits数据库,从上一阶段生成的MSA开始运行HHblits2,分别生成MSA-2和MSA-3。剩下的四个MSA通过qMSA过程生成,使用HHblits2在Uniclust30数据库(版本2020_01)中搜索目标序列创建MSA-4。接下来,利用Jackhmmer、HHblits3和HMMsearch通过UniRef90、BFD和Mgnify数据库检测到的序列,构建自定义的HHblits数据库,利用HHblits2从前一阶段生成的MSA开始搜索,分别创建MSA-5、MSA-6和MSA-7。为了选择最终的MSA,从7个MSA开始运行一个快速TripletRes接触映射预测,具有最高累积概率的MSA将作为最终的MSA。
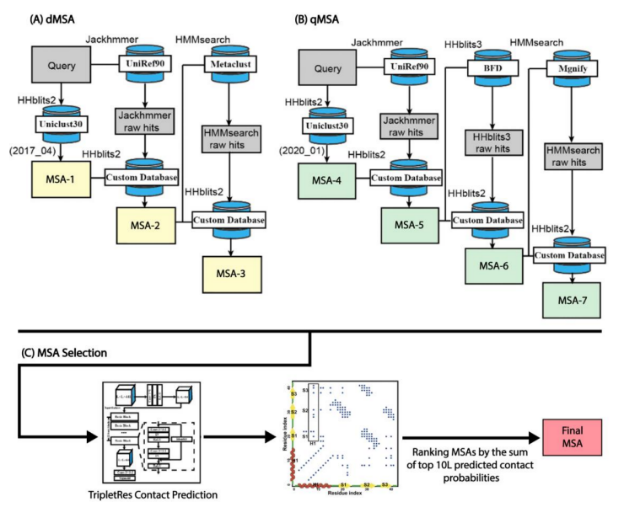
图2.DeepMSA2流程
DeepPotential空间约束预测**:**
从选定的MSA开始,提取两组一维和二维特征。二维特征包括通过伪似然最大化(PLM) 得到的22-state Potts模型的原始耦合参数和原始互信息(MI)矩阵,其中Potts模型的22个状态代表20个标准氨基酸、一个非标准氨基酸类型和一个gap状态。在DeepPotential中,CCMpred被用来拟合Potts模型。PLM和MI矩阵中每个残基对的对应参数被提取为额外的特征,用于测量MSA中特定目标的协同进化信息。一维特征包含Potts模型字段参数、隐马尔可夫模型(HMM)特征和自互信息,以及MSA的one-hot编码和其他描述符。接下来,这些一维和二维特征分别被输入残差网络,其中每个特征分别通过一组一维和二维残差块,然后平铺在一起。平铺后的特征输入另一个残差网络,该神经网络输出残基间的相互作用项,包括Cα-Cα距离、Cβ-Cβ距离和残基间的方向。预测的空间约束用对应特定距离/角度值的bin表示, DeepPotential预测空间约束落在特定bin内的概率。DeepPotential模型的训练对象是从PDB收集的26151个非冗余蛋白。
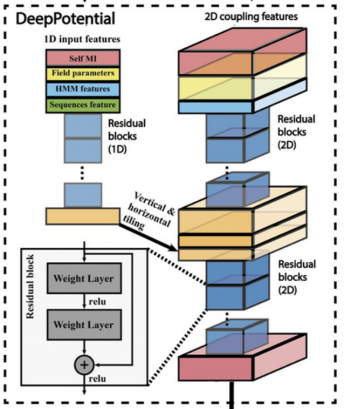
图3.DeepPotential预测空间约束流程
DeepFold Force Field**:**
DeepFold能量函数是以下各项的线性组合: 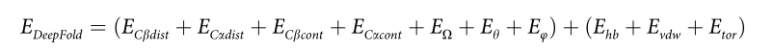
L-BFGS折叠模拟**:**
在DeepFold中,蛋白质结构由其主链原子(N, H, Cα,C和O)、Cβ原子和侧链质心确定。初始构象是由ANGLOR通过一个小的神经网络预测的骨干扭转角(φ,ψ)生成的。采用L-BFGS进行构象搜索模拟,对骨架扭转角进行优化。L-BFGS是一种基于梯度下降的优化方法。在每一步k处,模拟的搜索方向dk由下式确定:
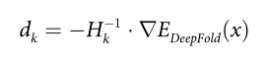
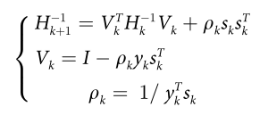
骨干扭转角(φ,ψ)的更新如下式:
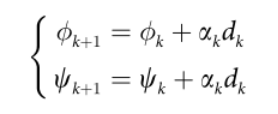
αk的值由Armijo线搜索技术确定,并决定了沿给定搜索方向移动的范围。在DeepFold中,最多执行10次L-BFGS迭代,每次迭代有2000个步骤,或者直到模拟收敛。最终的模型选择在折叠模拟过程中产生的能量最低的模型。
**3.**结果与讨论
距离和方向约束对全局折叠精度有主要影响:
为了测试DeepFold,文章从SCOPe 2.06数据库中收集了221个非冗余(序列一致性<30%)蛋白质以及CASP 9-12中的FM目标。为了检验DeepFold能量函数不同成分的重要性,使用来自DeepPotential的不同空间约束组合对221个测试蛋白运行DeepFold,建模结果如下图。总体而言,仅使用物理能量函数(GE)的TM平均得分仅为0.184。TM评分>0.5时表示正确折叠,即预测模型和原生结构具有相同的全局折叠,这也就表明仅使用物理能量函数无法正确折叠任何测试蛋白。进一步加入Cα和Cβ接触约束后,TM得分提高到0.263,221个测试蛋白中有4个(1.8%)成功折叠。添加Cα和Cβ距离约束后,测试数据集的TM平均得分提高到0.677,提高了157.4%,其中76.0%的测试蛋白能够正确折叠。最后,残基间方向的加入进一步将平均TM评分提高到0.751,成功折叠蛋白比例为92.3%。总的来说,随着约束细节水平的增加,能量景观越来越平滑,因此L-BFGS折叠模拟产生的测试蛋白质的平均TM得分增加。
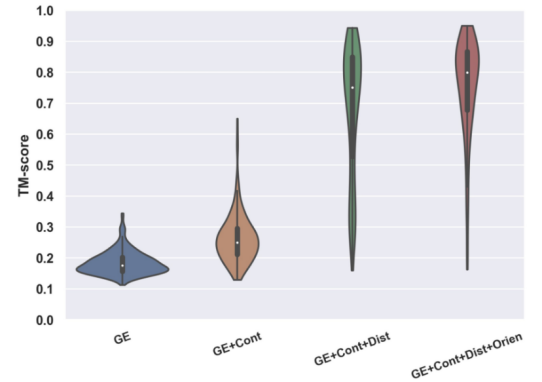
图4.不同的空间约束对DeepFold建模精度的贡献
物理能量函数提高了局部物理结构质量:
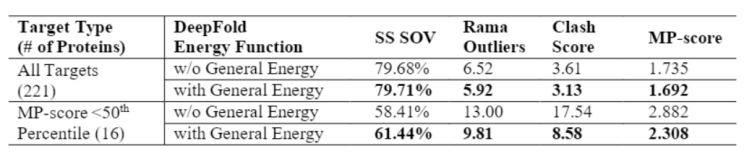
基于深度学习的约束预测精度的快速提高,使人们对物理能量函数在深度学习时代的作用产生了疑问。事实上,DeepFold准确性的主要贡献来自DeepPotential生成的大量准确预测约束,这些约束的加入极大地提高了平均TM得分。然而,物理能量函数解释了驱动蛋白质折叠的基本作用力,如氢键相互作用和范德华力,在提高预测模型的物理质量方面发挥了重要作用,尤其是当模型质量较差时。下表列出了使用和不使用GE时生成模型的几个质量指标。在加入GE函数后,MolProbity评分从1.735改善至1.692。在二级结构质量(SOV评分)、Ramachandran异常值的数量和空间冲突评分方面也观察到改善的趋势。最显著的改进体现在模型质量较差时的冲突评分上。表1.GE对DeepFold建模效果的作用
下图为一个来自SCOPe蛋白d1xsza2的案例研究,其中的模型在包含和不包含物理能量函数的情况下生成。在没有GE函数的模型中,有几个残基直接相互重叠,导致严重的空间冲突。这些冲突通过加入GE得到解决,得到的模型MolProbity分数降低到1.624(第92百分位),冲突分数低至1.2。显然,仅仅满足深度学习提供的几何约束可能会导致模型在物理上不现实,而引入物理能量项可能会一定程度上缓解这一问题。
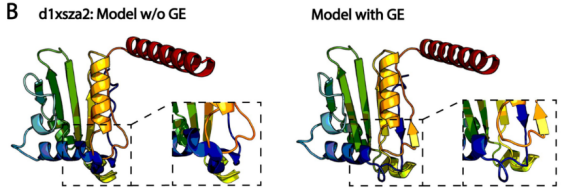
图5.d1xsza2案例研究 能量函数对于提高模型物理质量的重要性
DeepFold与其他领先建模方法的比较**:**
为了进一步评估DeepFold的性能,文章将221个测试蛋白的建模结果与领先的基于接触图的方法(C-I-TASSER)、基于距离(DMPfold)和基于距离方向(trRosetta)方法以及经典的I-TASSER进行了比较。为了公平比较,使用与DeepFold相同的MSA(由DeepMSA2生成)用于DMPfold、trRosetta和C-I-TASSER的深度学习约束预测,以及I-TASSER和C-I-TASSER中的LOMETS的模板识别。如表所示,221个测试蛋白上的DeepFold模型TM平均得分显著高于所有对照方法。例如,I-TASSER生成模型的平均TM得分仅为0.383,其中DeepFold获得的平均TM得分(0.751)比I-TASSER高96.1%。在C-I-TASSER中加入深度学习接触映射后,TM得分提高到0.584。尽管如此,DeepFold的TM平均得分仍比C-I-TASSER高28.6%。这主要是由于DeepFold同时利用了距离和方向约束,其中包含了比C-I-TASSER中使用的接触映射更详细的空间信息。中位数的结果与平均值相似,DeepFold的TMscore中位数为0.800,而I-TASSER和C-I-TASSER的TM得分中位数分别为0.357和0.607。 表2.不同方法在221个蛋白上的建模结果比较

DeepFold的表现也优于其他两种领先的基于距离(DMPfold)和基于距离方向(trRosetta)的方法,其中DMPfold的TM平均/中位数得分为0.657/0.710,trRosetta的TM平均/中位数得分为0.694/0.749,DeepFold的TM平均分/中位数比DMPfold高14.3%/12.7%,比trRosetta高8.2%/ 6.8%。此外,下图展示了DeepFold与不同方法的正面对比,在221个测试蛋白中,DeepFold分别在194和204个蛋白上优于trRosetta和DMPfold。与DMPfold相比,DeepFold的一个明显优势是利用残基间的二面角进行定向,这使DeepFold的TM评分大幅提高。与trRosetta相比,由于这两种方法都使用距离和方向约束,DeepFold的主要优势是DeepPotential生成的约束具有较高的精度。在AlphaFold提交模型的31个CASP13 FM靶标上比较了DeepFold和AlphaFold的建模精度。从下图E可以看出,在31个FM目标中,DeepFold在20个目标上的表现优于AlphaFold,平均而言,DeepFold的TM得分为0.636,而AlphaFold的TM得分为0.589。
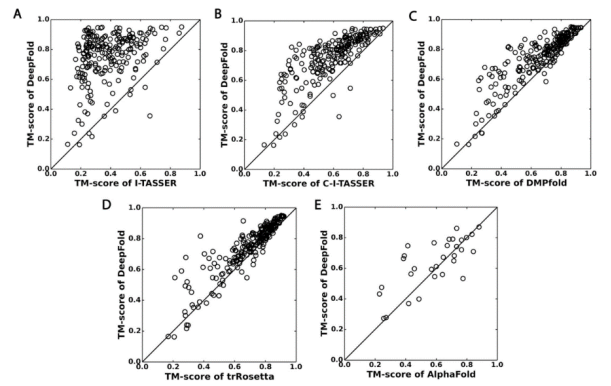
图6.DeepFold与其他蛋白质结构预测方法的TM得分对比
与传统的从头预测方法相比,DeepFold大大提高了蛋白质折叠的准确性和速度:
Rosetta和QUARK是两种最有影响力的碎片组装方法,在以往的CASP实验中一直被列为从头预测蛋白质结构的首选方法。然而,传统从头预测方法的一个主要缺点是随着蛋白质长度的增加,其建模性能下降,这使得它们在建模超过150个残基组成的蛋白质结构时明显不可靠。为了检验深度学习对长蛋白质序列从头预测的影响,文章将DeepFold与Rosetta和QUARK进行了比较,下图B描述了DeepFold、QUARK和Rosetta的TM评分与蛋白质长度的关系。数据显示,随着蛋白质长度的增加,DeepFold的性能保持一致,其中由350-450个残基组成的大型蛋白质的平均TM评分实际上高于长度<150个残基的小蛋白质(0.809 vs. 0.742),这主要是由于为较大的蛋白质集收集到更有利的MSA。然而,随着蛋白质长度的增加,QUARK和Rosetta的性能明显下降;长度小于150个残基的蛋白质,夸克和Rosetta的平均TM得分分别为0.329和0.304,而长度在350 - 450个残基之间的蛋白质,夸克和Rosetta的平均TM得分分别为0.190和0.196。从这些结果来看,DeepFold在整个数据集上的表现明显优于QUARK和Rosetta,特别是在数据集中最长的蛋白质上,DeepFold的TM平均得分比QUARK高325.8%,比Rosetta高312.8%。碎片组装方法的另一个主要限制是,它们需要长时间的模拟来充分探索可用的巨大结构空间。在图A中列出了对于不同蛋白质长度的DeepFold和QUARK、trRosetta的折叠时间需求的比较。结果表明,DeepFold的速度比QUARK快了好几个数量级,特别是对于大蛋白。DeepFold在测试集上的平均运行时间为6.98分钟,而QUARK的平均运行时间为1830.82分钟,平均蛋白质长度为188.1个残基。这说明QUARK对于一个模拟折叠需要的计算时间是DeepFold的262.3倍,并且随着序列长度的增加,差异更大。DeepFold的运行时间与trRosetta相似,在测试数据集上构建模型平均需要5.48分钟。尤其重要的是,对于更大的蛋白质,大大减少的折叠时间并没有导致模型质量恶化,这证明了深度学习约束能够有效地平滑能量景观,从而实现跨蛋白质长度的快速和精确优化。
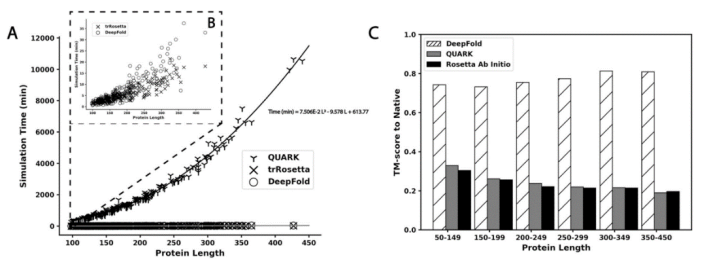
图7.模拟时间和TM评分与蛋白质长度的相关性
**3.**结论
DeepFold首先使用DeepMSA2在多个全基因组和宏基因组数据库中搜索目标序列,创建MSA。之后从得到的MSA中提取协同进化耦合矩阵,输入DeepPotential的ResNet中用于预测空间约束,包括距离/接触映射和残基间方向。然后将这些约束转换为基于深度学习的势能,该势能与物理势能一起指导L-BFGS折叠,最后生成完整的模型。在221个目标测试集上,DeepFold显著优于其他从头预测方法,如Rosetta、QUARK、I-TASSER、C-I-TASSER、DMPfold和trRosetta,而在CASP13 FM目标测试集上,DeepFold优于AlphaFold。在基准数据集上,Rosetta、QUARK和I-TASSER分别只能正确折叠0.9%、2.7%和24.0%的蛋白质,而DeepFold成功折叠了92.3%的测试蛋白质,平均TM得分为0.751,Rosetta、QUARK和I-TASSER分别为0.260、0.274和0.383。此外,DeepFold的TM平均得分比其他基于深度学习的领先方法DMPfold和trRosetta分别高出7.8%和13.9%,而这些方法都是基于相同的MSA。除了提高精度外,DeepFold的运行时间与其他基于梯度下降的方法(如trRosetta)相似,但比传统的基于碎片组装的方法快200多倍。DeepFold的成功主要是因为它将基于知识的势能与大量精确预测的空间约束有效结合起来,这有助于平滑能量景观,使L-BFGS优化变得容易。