蛋白质是维持生命系统的必要大分子,通过在结合位点处与其他分子的相互作用来发挥生理功能。这些结合区域的确证和表征对于确定蛋白质功能具有重要意义,是药物设计和发现等过程中的一个基本步骤。然而,通过实验检测蛋白质-配体结合位点是费时和昂贵的。因此,作者提出了GRaSP-web,一个基于GRaSP(基于图的残基邻域策略预测结合位点)方法的web服务器。GRaSP是一种以残基为中心的方法,使用机器学习来预测潜在的配体结合位点残基。GRaSP-web可从https://grasp.ufv.br免费访问。研究背景
目前,大量蛋白质的生物学功能仍是未知的,对结合位点的了解,以及参与配体结合的氨基酸残基的鉴定,是蛋白质功能表征的关键步骤。研究人员已开发了一系列基于结构的方法来识别蛋白质结合位点:例如Firestar,基于与FireDB数据库中已知配体结合残基的局部序列保守匹配来识别配体结合残基;FunFold3,在目标模型上执行包含相关配体的结构模板的叠加,来识别结合残基;COACH,一种共识方法,结合自己内部开发的算法TM-SITE和S-SITE,以及第三方预测工具COF ACTOR、FINDSITE和ConCavity,预测单链结构的结合位点;LigDig,结合配体相互作用网络的数据库来识别相似的配体及其结合蛋白,从而指出潜在的蛋白-配体结合位点;GASS,提出了一种从CSA中搜索未知蛋白质中活性位点结构模板的遗传算法。但这些方法普遍存在执行时间较长,无法预测蛋白质界面处的结合位点,以及缺乏可解释的和可视化的结果等缺陷[1]。 GRaSP在原子水平上将特定残基及其邻域表示为图来感知残基环境,并使用监督学习来预测结合位点处的氨基酸残疾。与其他6种以残基为中心的方法相比,GRaSP表现出相当或更优的预测性能。************模型与方法****************1.模型框架
GRaSP将预测配体结合位点的残基问题建模为二元分类任务,其目的是预测每个残基是否在结合位点上。监督学习策略使用数据矩阵 G 进行训练,其中每一行表示一个残基 r,每一列编码一个描述符。因此,每个残基 r 被编码为一个特征向量,共由42个描述符构成;整个蛋白质结构 P 则被编码为一组特征向量。GRaSP采用了一种以残基为中心的策略,将每个残基和它相邻的前两层建模为一个邻域图,以感知残基局部物理化学环境信息。对于蛋白质的每个残基,GRaSP将其编码为数值描述符,,其中其原子和相互作用的拓扑性质和物理化学性质被表示为一个图,然后该图又被编码为一个特征向量。该特征向量集用于训练预测模型。GRaSP使用极端随机树算法执行监督学习任务,该算法属于一类集成分类器,在scikit-learn(scikit-learn.org)中实现。2.服务器
图1显示了GRaSP-web处理步骤。将输入蛋白质的每个残基建模为邻域图,并将邻域图编码为特征向量。然后,从BioLip数据库中搜索与输入蛋白质相似模板构建一个训练数据集。接下来,使用平衡分类器的集合来预测输入蛋白的结合位点的残基。最后,在molecular viewer中显示预测的结合位点残基以及每个预测的置信度分数。残基也可以聚集在结合位点上,并为每个位点提供潜在的配体;同时,作者还提出了描述符的相对重要性,以支持用户对预测的理解。3.输入
为了进行预测,用户可以上传一个或多个PDB格式的三维结构,或提供PDB识别码(图1A),GRaSP-web将从PDB数据库中检索并存储相应的结构。当蛋白提交后,GRaSP使用图形建模来感知每个残基的化学环境,并将其编码为数值特征(图1B,C),这些特征向量被组合成一个矩阵用于结合位点预测(图1C)。查询结构的氨基酸序列与来自BioLip的配体结合模板进行匹配。将这些模板作为训练数据来构建监督机器学习模型。 数据不平衡是预测结合位点的固有问题。非结合位点残基的数量通常大于结合位点残基的数量,这降低了传统分类器的预测能力。因此,GRaSP-web将重采样方法和集成树相结合,以平衡数据和改进预测(图1E)。GRaSP-web处理含有300个氨基酸残基的蛋白质复合物大约需要20秒。
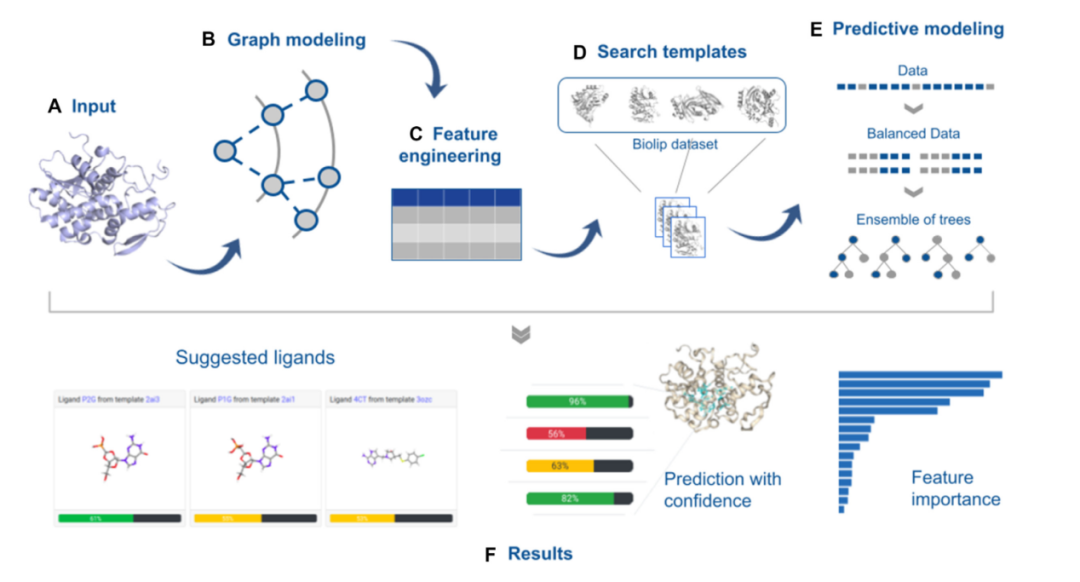
图1** GRaSP-web工作流程图********[**********1]
4.输出
GRaSP-web会为每个提交的任务分配一个URL,以便用户访问结果或跟踪任务状态。每个提交的蛋白的标准输出包括一个预测的配体结合位点残基列表(图2A),可以下载到本地计算机上。每个预测的残差都有一个置信度得分,用来表示其可靠性。此外,GRaSP-web使用NGL viewer (nglviewer.org)进行分子可视化,查询的蛋白会被显示出来,结合位点残基会被高亮显示(图2B)。 为了展示以口袋为中心的视角,作者提供了基于密度的DBSCAN方法来将预测的残基聚类,以模拟结合口袋 (图2C)。随后,使用PocketMatch算法将每个残基簇与用作训练数据集的整个结合位点数据库进行比较,相似的结合位点往往会与相似的配体结合,由此可以预测哪些配体有可能与一个残基簇结合。GRaSP-web为每个候选配体提供了一个评分条,它代表了从预测残基簇和模板数据库结合位点之间的PocketMatch得分。

图2 GRaSP-web结果页面********[********************1]************结果与讨论
1.评估GRaSP-web的性能
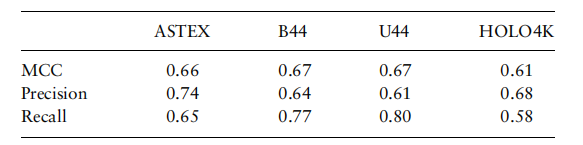
作者使用多种数据集来评估GRaSP-web的性能,这些数据集捕捉了蛋白质结构的不同特征,包括一些类药物复合物、结合/非结合态蛋白质和多聚结构。评估方法的预测性能采用马修斯相关系数(MCC)、精度、召回率、结合距离测试(BDT)和口袋中心与任何配体原子之间的距离(DCA)等指标来衡量。 如表1 所示,作者对ASTEX数据集中的85个蛋白质进行了测试,MCC为0.66,精度达到了0.74,这对于残基结合位点来说是一个很好的表达值,意味着假阳性的发生率较低,这对于可药结合位点的获得非常重要,可避免了在非结合的阴性区域进行不必要的对接实验。接下来,使用B44/U44数据集对44个结合和非结合区域进行GRaSP性能比较,该结构包含在结合和非结合状态下完全相同的蛋白质结构。GRaSP在两种状态下取得了非常相似的结果,MCC为0.67。最后,作者使用GRaSP指出HOLO4K中4543个多链结构的结合位点残基,MCC 达到了0.61;而最先进的方法只能处理单链结构,并在其500个单链蛋白的基准数据集中MCC也只达到了0.60[2]。 表1 针对不同数据集的GRaSP结果**********[******2]
2.与现有模型的比较****2.1以残基为中心的方法比较
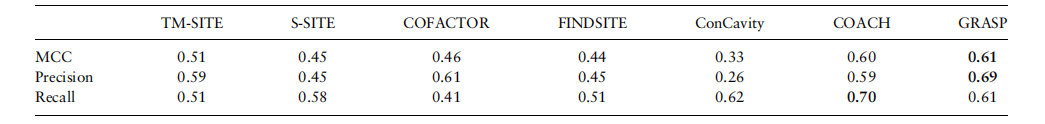
COACH是一种基于序列和结构的方法的混合方法,同时也是目前最先进的以残基为中心的方法。作者使用来自COACH的基准数据集将GRaSP方法与COACH方法进行比较,数据集包含500个非冗余单链蛋白。GRaSP的MCC为0.61,仅需10到20秒即可完成预测,而COACH的MCC为0.60,需要2-5小时来预测类似大小的蛋白质。MCC是衡量二分类模型结果的评估指标之一,也是COACH作者用来与其他方法比较的主要指标,比单独使用的精确度和召回率更加均衡。表2总结了测试结果。表2 GRaSP方法和coach方法的比较结果************[****2]
2.2以口袋为中心的方法比较
在以口袋为中心的预测中,常用的方法有Fpocket, metapoket和Sitehound, P2Rank 和DeepSite等,后二者是口袋中心类别中当前最先进的方法。利用DBSCAN 对GRASP预测为阳性的残基集进行聚类,当预测为阳性的残基形成一个高密度区域时,该区域被认为是一个口袋。使用DCA指标评估口袋预测因子,这个参数代表了从预测口袋中心到配体任何原子的距离。给定一个具有n个结合位点的蛋白质,对于每一种考虑的方法,作者进行了n次口袋预测。如果其相应配体与给定方法的n次预测中的任何一个之间的最小距离低于阈值,则认为该结合位点被正确预测。图3显示了预测器的成功率,其计算方法为为正确预测的结合位点数量除以结合位点总数。GRaSP仅位于P2Rank之后,这个结果是令人惊喜的,因为GRaSP最初并不是为了预测潜在的口袋而设计的。
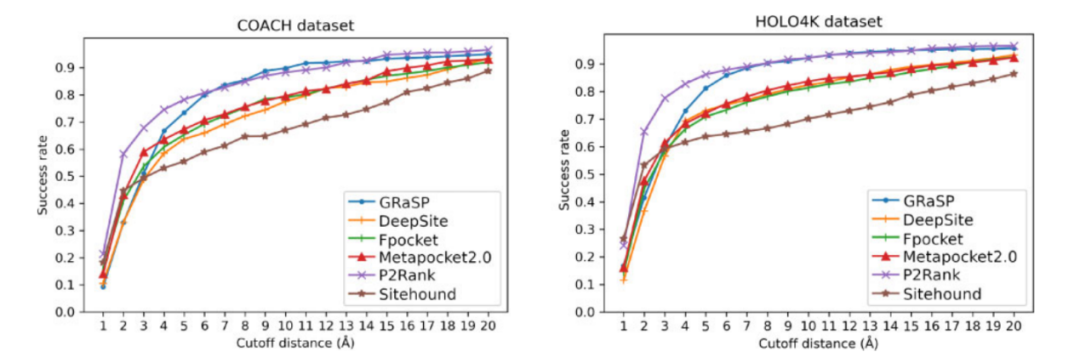
图3 GRaSP与口袋中心法的预测性能比较**************[**2]
在预测结合位点残基时,需要强调的另一个方面是蛋白质的构象状态。许多蛋白质在与配体分子结合时会发生诱导契合,从而变蛋白质的构象。结合位点预测方法应该忽略这些蛋白质构象的变化,只关注结合态和非结合态空腔的物理化学模式。作者使用了一个包含两种状态下44个蛋白质结构的数据集来评估GRaSP-web是否能够正确预测结合位点残基。图4显示了HIV蛋白酶与VAC配体(pdb 4PHV),以及非结合状态的HIV蛋白酶(pdb 3PHV)的结构。GRaSP-web能够正确预测两种情况下的结合位点残基,实现了结合/非结合态的总体平均MCC值0.67。
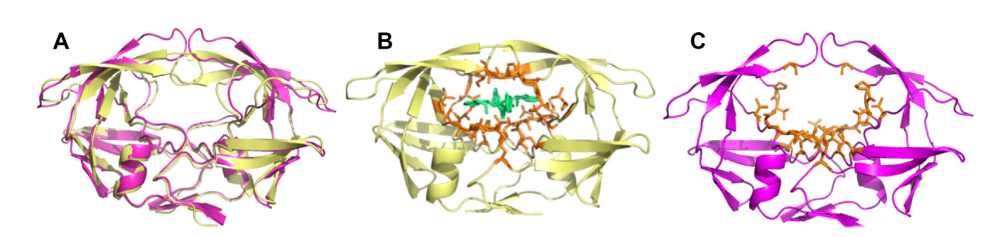
图4 HIV蛋白酶的蛋白质结构****************[********************1]********************总结
在这项工作中,作者提出了一种新的、以残基为中心的、基于图的可扩展的方法来预测配体结合位点残基,并建立了网络计算服务器GRaSP-web。GRaSP-web提供了一个用户友好的界面,可对预测结果进行可视化。用户可以提交自己的蛋白质结构,或者PDB数据库中的结构,对结合位点残基进行预测。在多个基准测试中进行的实验表明,此服务器能够预测结合位点残基,在速度和精确度等方面与现有方法相近或优于现有方法。
**参考文献 **
[1] Santana C A , Izidoro S C , Demelo-Minardi R C , et al. GRaSP-web: a machine learning strategy to predict binding sites based on residue neighborhood graphs[J]. Nucleic Acids Research, 2022, 50(W1):W392-W397. doi:10.1093/nar/gkac323. [2] Moraes J P A , Thornton J M , Borkakoti N , et al. GRaSP: a graph-based residue neighborhood strategy to predict binding sites[J]. Bioinformatics, 2020, 36(Suppl_2):i726-i734. doi:10.1093/bioinformatics/btaa805.
供稿:彭丹妮
校稿:刁妍妍/沈子豪编辑:毛丽韫华东理工大学/上海市新药设计重点实验室/李洪林教授课题组▼招聘博后▼华东理工大学李洪林教授团队诚聘博士后


Li's Lab地址:上海市梅陇路130号 电话:021-64250213课题组网站:http://www.lilab-ecust.cn 长按扫码可关注