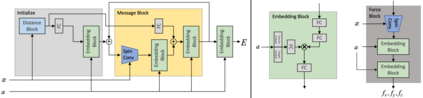
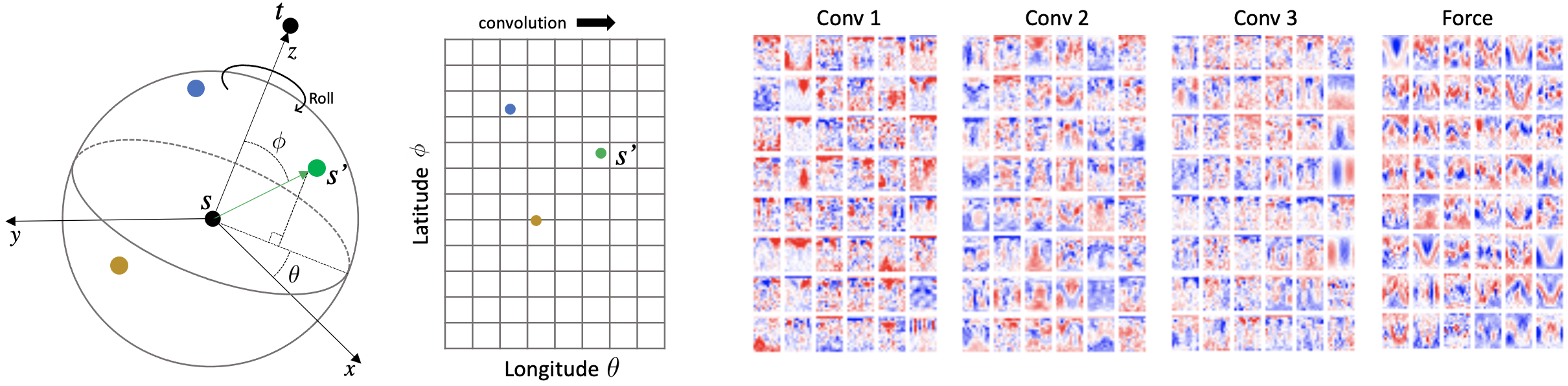
Progress towards the energy breakthroughs needed to combat climate change can be significantly accelerated through the efficient simulation of atomic systems. Simulation techniques based on first principles, such as Density Functional Theory (DFT), are limited in their practical use due to their high computational expense. Machine learning approaches have the potential to approximate DFT in a computationally efficient manner, which could dramatically increase the impact of computational simulations on real-world problems. Approximating DFT poses several challenges. These include accurately modeling the subtle changes in the relative positions and angles between atoms, and enforcing constraints such as rotation invariance or energy conservation. We introduce a novel approach to modeling angular information between sets of neighboring atoms in a graph neural network. Rotation invariance is achieved for the network's edge messages through the use of a per-edge local coordinate frame and a novel spin convolution over the remaining degree of freedom. Two model variants are proposed for the applications of structure relaxation and molecular dynamics. State-of-the-art results are demonstrated on the large-scale Open Catalyst 2020 dataset. Comparisons are also performed on the MD17 and QM9 datasets.
翻译:通过高效模拟原子系统,可以大大加快在应对气候变化所需的能源突破方面的进展。基于第一原则的模拟技术,如密度功能理论(DFT),由于计算成本高,其实际使用有限。机器学习方法有可能以计算效率高的方式接近DFT,这可能会大大增加计算模拟对现实世界问题的影响。接近DFT带来若干挑战。其中包括精确模拟原子之间相对位置和角度的微妙变化,以及实施轮用变换或节能等限制。我们采用了一种新的方法,在图形神经网络中模拟相邻原子之间的三角信息。通过使用一个对称的地方协调框架和对剩余自由程度进行新的旋转,实现网络优势信息的旋转。为结构放松和分子动态的应用提出了两个模型变量。在大规模开放的2020年卡塔利数据集中展示了国家艺术成果。还进行了MDD17和MQ数据集的比较。