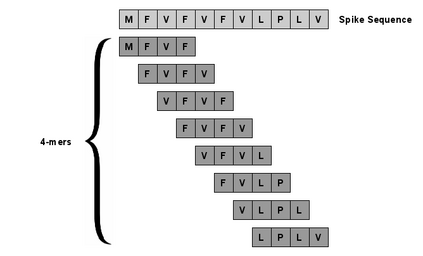
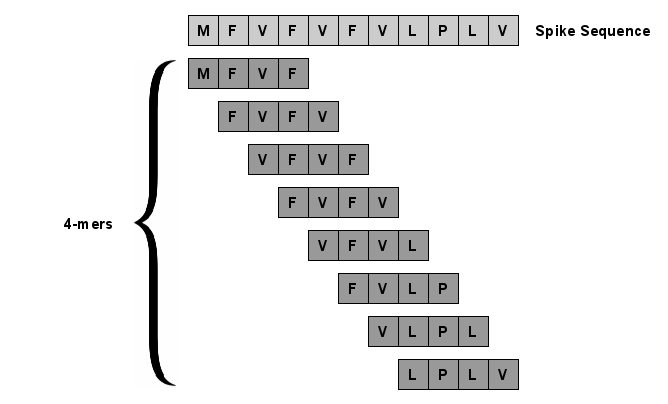
With the rapid spread of the novel coronavirus (COVID-19) across the globe and its continuous mutation, it is of pivotal importance to design a system to identify different known (and unknown) variants of SARS-CoV-2. Identifying particular variants helps to understand and model their spread patterns, design effective mitigation strategies, and prevent future outbreaks. It also plays a crucial role in studying the efficacy of known vaccines against each variant and modeling the likelihood of breakthrough infections. It is well known that the spike protein contains most of the information/variation pertaining to coronavirus variants. In this paper, we use spike sequences to classify different variants of the coronavirus in humans. We show that preserving the order of the amino acids helps the underlying classifiers to achieve better performance. We also show that we can train our model to outperform the baseline algorithms using only a small number of training samples ($1\%$ of the data). Finally, we show the importance of the different amino acids which play a key role in identifying variants and how they coincide with those reported by the USA's Centers for Disease Control and Prevention (CDC).
翻译:随着新颖的冠状病毒(COVID-19)在全球的迅速传播及其持续突变,设计一个系统以查明SARS-COV-2的不同已知(和未知)变异体至关重要。确定特定变异体有助于理解和模拟其扩散模式,设计有效的缓解战略,防止今后爆发。它还在研究已知疫苗对每种变异的功效和模拟突破性感染的可能性方面发挥着关键作用。众所周知,峰值蛋白包含了与corona病毒变异体有关的大多数信息/变异。在本文件中,我们使用峰值序列对人类的冠状病毒的不同变异进行分类。我们表明,保持氨基酸的顺序有助于基本分类者取得更好的性能。我们还表明,我们可以用少量的培训样本来训练我们的模型,以超越基线算法。最后,我们展示了不同的氨基酸的重要性,它们在确定变异体方面起着关键作用,以及它们如何与美国疾病控制和预防中心所报告的那些变异体(CD C)相一致。