利用Transformer替代MSA从蛋白序列中学习Contact Map
点击上方“CVer”,选择加"星标"置顶
重磅干货,第一时间送达

本文转载自:GoDesign
-
对目标蛋白及相关序列进行多重序列比对,实现这一步的常用工具包括Jackhmmer和HHblits; -
根据序列比对结果,对残基间的进化相关性进行建模。实现这一步的典型做法是训练马尔可夫随机场(Markov Random Field, MRF),如Gremlin[2]; -
根据共进化信息对蛋白结构进行优化。可行的方法包括:将共进化对作为距离约束引入已有的优化器[3],或将其作为特征引入深度学习模型中[1]。
-
ESM-1b能够绕开MSA流程里许多繁重的计算,如序列检索、序列匹配、MRF模型的训练等,而仅需一次网络前传(forward pass)就能得到结果; -
ESM-1b具有很好的泛化能力,可以用于任意序列,而不像MSA要求有足够多的同源数据。
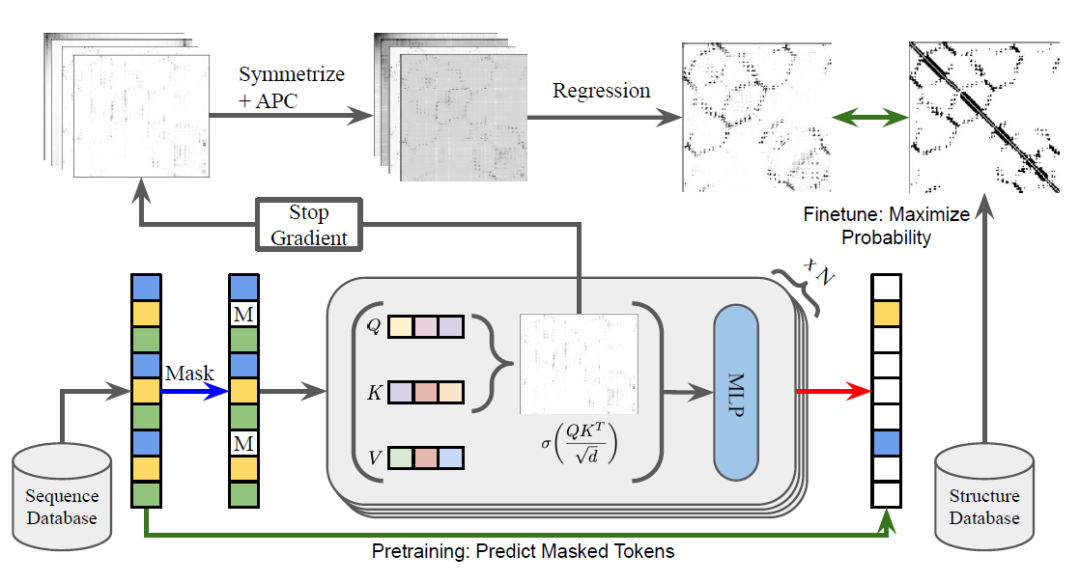
图1:ESM-1b的训练流程。
我们注意到在上述流程中,除了最后的逻辑斯蒂分类以外,ESM-1b主体部分(即Transformer)的训练不依赖三维结构信息,仅需未标注的蛋白序列信息,因此该流程在很大程度上是无监督的。ESM-1b模型在无监督预训练时使用了UniRef50数据集[4],在模型评价时选择了trRosetta的训练数据库[5],包含15051个蛋白,经过处理后蛋白数为14882,其中20个数据点用于逻辑斯蒂分类器的训练,其他用于模型评价。
——方法评价——
ESM-1b的主要比较对象是Gremlin,属于经典的基于MSA的方法。除此之外,作者还将ESM-1b与其他基于Transformer的方法进行了对比,包括TAPE[6],ProtBert-BFD[7]以及ESM-1(即ESM-1b的上一个版本,ESM-1b相比第一版进行了诸多超参数和构架上的优化)。模型评价的主要指标是Contact Map的预测准确性,分为短程(6<=sep<12)中程(12<=sep<24)以及长程(sep>=24)的残基接触预测准确性。评价结果如表1所示。作者还修改了模型的不同超参数(在下文中具体讨论),结果如表2所示。
表1:不同模型在Contact Map预测任务上的表现
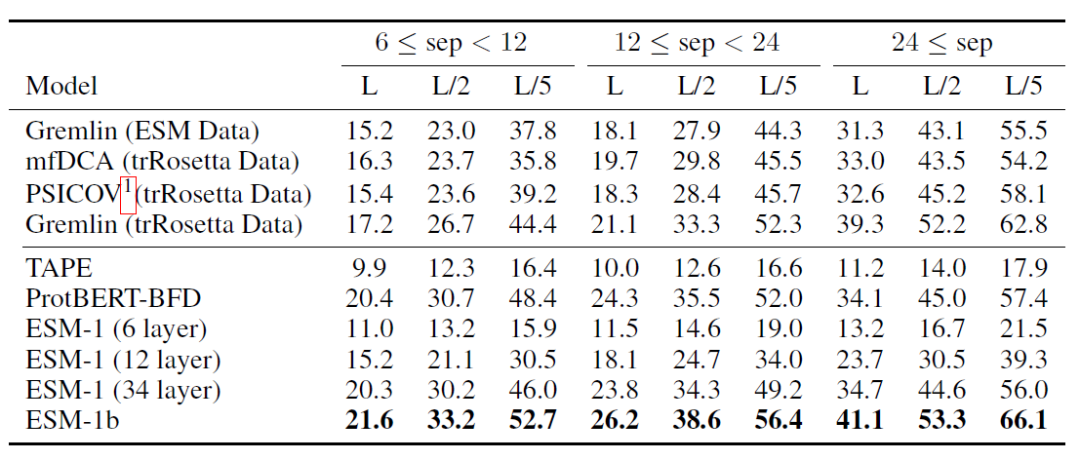
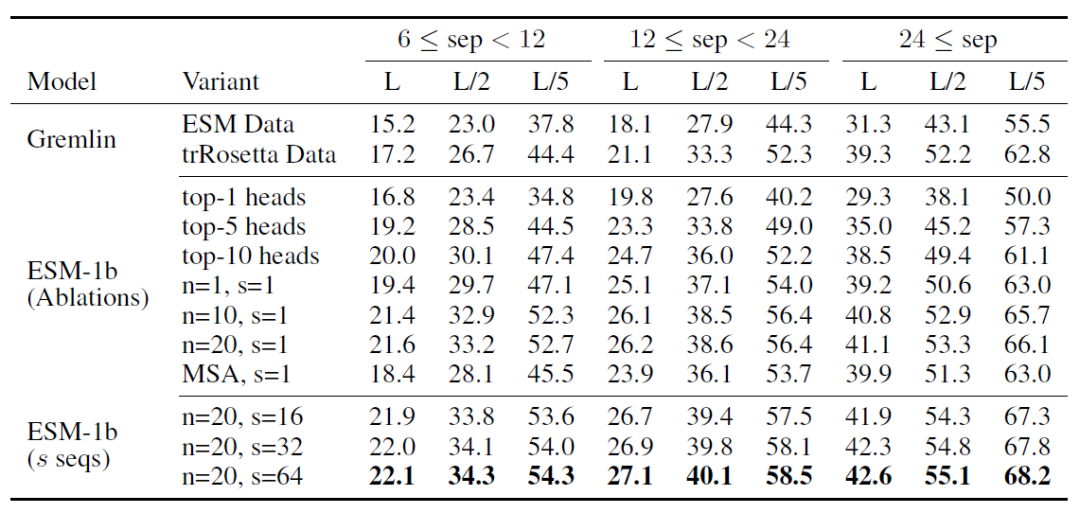
表2:不同超参数对于模型表现的影响
从结果上来看,ESM-1b在各项指标上均优于其他模型。具体而言,ESM-1b有如下特点:
与其他Transformer方法的对比:ESM-1b相比TAPE、ProtBERT-BFD以及ESM-1,在相互作用预测上表现都要更好(表1:第5-10行);
与经典方法(Gremlin)的比较:作者发现ESM-1b在长程、中程和短程的相互作用预测上相比Gremlin都更加准确(表1第1、4行与最后一行),说明单独使用ESM-1b在蛋白结构预测上已经展现了较好的潜力。
与经典方法的结合:作者还考虑了将ESM-1b与MSA结合构建ensemble model。具体的做法是:将MSA比对结果排名靠前的序列挑选出来,利用模型预测contact map,最后将结果进行平均。作者发现ensemble model能够进一步提升模型表现(见表2中s=16,32,64,其中s为用到的MSA序列数);
无监督预训练对模型准确性的贡献:通过分析结果,作者发现ESM-1b的优异表现主要来自于模型的无监督预训练(即Transformer)。作者首先发现,仅使用attention map中的一个attention head就可以达到与Gremlin同样水平的表现(表2,top-1 heads),说明逻辑斯蒂监督学习得到的权重在模型表现上并非主要贡献。其次,作者发现逻辑斯蒂分类仅需很少的训练样本(表2第6-8行,样本数n=1, 10, 10),就可以实现优于Gremlin的表现,进一步说明了无监督训练的重要作用。
——小结——
总结起来,Rao等人提出了一种基于Transformer的蛋白序列无监督学习方法。和简单的逻辑斯蒂分类结合之后,该模型在contact map预测上能够达到比基于MSA的方法更好的表现。这一类方法相比MSA有许多潜在的优势,例如不需要进行序列比对、运行更方便、具有很好的泛化能力等,因此在蛋白结构预测任务上有潜力成为MSA的替代方法。但是这篇文章并没有回答所有问题。例如,这篇文章只关注了contact map预测,而并没有实际考察其在实际三维结构预测方面的表现。因为缺乏这方面的结果,目前我们暂不明确这一类模型在多大程度上能够替代MSA。
参考文献:
[Main] Rao, Roshan, et al. "Transformer protein language models are unsupervised structure learners." bioRxiv (2020).
[1] Senior, Andrew W. et al. “Improved Protein Structure Prediction Using Potentials from Deep Learning.” Nature 577.7792 (2020): 706–710.
[2]Balakrishnan, Sivaraman et al. “Learning Generative Models for Protein Fold Families.” Proteins: Structure, Function, and Bioinformatics 79.4(2011): 1061–1078.
[3]Ovchinnikov, Sergey et al. “Improved de Novo Structure Prediction in CASP11 by Incorporating Coevolution Information into Rosetta.” Proteins: Structure,Function, and Bioinformatics 84.S1 (2016): 67–75.
[4]Suzek, Baris E., et al. "UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches." Bioinformatics 31.6 (2015): 926-932.
[5]Yang, Jianyi, et al. "Improved protein structure prediction using predicted interresidue orientations." Proceedings of the National Academy of Sciences 117.3 (2020): 1496-1503.
[6]Rao, Roshan, et al. "Evaluating protein transfer learning with tape." Advances in Neural Information Processing Systems 32 (2019): 9689.
[7] Elnaggar, Ahmed, et al. "ProtTrans: Towards Cracking the Language of Life's Code Through Self-Supervised Deep Learning and High-Performance Computing." arXiv:2007.06225 (2020).
点击左下角的"阅读原文"即可查看原文章。
作者:李亦博 | 审稿:徐优俊
王世伟 | 编辑:林康杰
CV资源下载
后台回复:Transformer综述,即可下载两个最新的视觉Transformer综述PDF,肝起来!
重磅!CVer-Transformer交流群成立
扫码添加CVer助手,可申请加入CVer-Transformer方向 微信交流群,也可申请加入CVer大群,细分方向已涵盖:目标检测、图像分割、目标跟踪、人脸检测&识别、OCR、姿态估计、超分辨率、SLAM、医疗影像、Re-ID、GAN、NAS、深度估计、自动驾驶、强化学习、车道线检测、模型剪枝&压缩、去噪、去雾、去雨、风格迁移、遥感图像、行为识别、视频理解、图像融合、图像检索、论文投稿&交流、Transformer、PyTorch和TensorFlow等群。
一定要备注:研究方向+地点+学校/公司+昵称(如Transformer+上海+上交+卡卡),根据格式备注,才能通过且邀请进群

▲长按加微信群

▲长按关注CVer公众号
整理不易,请给CVer点赞和在看!