
2022年9月12日,哈尔滨医科大学生物信息科学与技术学院程亮教授团队和南京医科大学生物医学工程与信息学院刘云教授团队合作在Bioinformatics上发表文章《MGPLI: Exploring multigranular representations for protein-ligand interaction prediction》。作者提出了一个多粒度的蛋白-配体相互作用预测模型MGPLI。模型利用Transformer和CNN网络分别提取了蛋白和药物的子序列级和字符级信息,并将两者融合用于预测蛋白-配体相互作用。模型在不同的数据集上进行了评估,性能表现优秀。
1 摘要
目标:预测针对蛋白质-靶标的潜在药物结合亲和力的能力一直是计算药物发现的基本挑战。传统的体外和体内实验成本高且耗时,需要在大的化合物空间上进行搜索。近年来,基于深度学习的药物-靶标结合亲和力(drug-target binding affinity, DTA)预测模型取得了显著成功。
结果:随着Transformer模型最近的成功,作者提出了一种多粒度蛋白质-配体相互作用(Multi-Granularity Protein Ligand Interaction, MGPLI)预测模型,该模型采用Transformer编码器来表示字符级(character-level)特征和片段级(fragment-level)特征,建模残基和原子或其片段之间可能的相互作用。此外,作者使用卷积神经网络(CNN)提取基于transformer编码器输出的高级特征,并使用highway层融合蛋白质和药物特征。作者在不同的蛋白质-配体相互作用数据集上评估MGPLI,与最先进的基线相比,本模型表现出更好的预测性能。
2 问题形式化可以将DTI预测视为一个回归任务,即预测给定药物-靶标蛋白对之间的亲和力.药物的SMILES序列可以表示为:.蛋白的氨基酸序列可以表示为:.则,数据集中的样本可以表示为一个元组:
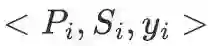
模型的目标则是通过建立一个从
到
的映射来预测药物-靶标蛋白之间的亲和力。
3 方法如图1所示,论文的主要步骤为:
- Tokenization:划分SMILES和氨基酸序列为字符级别和子序列级别的token序列。
- Token Embedding + Position Embedding,将1中划分的token嵌入向量,且为每个token添加位置嵌入,将两者之和作为token的最终嵌入。
- Transformer:利用Transformer对Token的嵌入进行编码,分别学习到药物和靶标蛋白不同粒度的高级特征。
- CNN:将3中的高级特征进行全局最大池化(global max pooling)进一步提炼特征,得到不同粒度的特征向量(Multi-grained feature vectors)。
- HighWay Layer:采用highway层融合4中得到的特征。
- Multi-Layer Perceptron:使用三个全连接层来预测结合亲和力。
下面详细介绍部分模块。
图1 MGPLI的模型架构
3.1 Tokenization
作者采用了名为sentence piece tokenization的算法对药物和蛋白序列进行片段化。sentence piece tokenization是一种数据驱动的算法,可以直接使用原始序列数据进行训练,并分词。对于蛋白,作者在来自于UniprotKB数据库的0.56M蛋白序列上进行了预训练;对于药物,作者在来自于ChEMBL数据库的1.6M化合物上进行了预训练。 作者将药物和蛋白分别进行子序列化和字符化,这是两种不同粒度的特征,可以互补。对于蛋白的氨基酸序列:,可以转化为子序列的序列:,和字符序列:.对于药物的SMILES序列:可以转化为子序列的序列:,和字符序列:. 3.2 多粒度嵌入(Multigranular embedding)对于得到的四种序列,作者首先将其通过四个可训练的矩阵, , , 进行嵌入。为了建模不同位置元件的关系,作者加入了绝对位置编码(absolute positional encoding):为了避免过拟合和不稳定的训练过程,作者添加了层标准化技术: 和dropout技术。综上可以将四种序列信息表示为,即
**3.3 Transformer-CNN **Transformer-CNN的结构如图2所示。transformer-CNN的优势是可以同时提取对预测DTA重要的全局序列信息和局部特征信息。
图2 提出的transformer-CNN的结构如右图所示,transformer-CNN单元如左图所示对于具体的transformer编码器,自然包括多头注意力层(multihead attention layer)、位置前馈网络(positionwise feedforward network)、残差连接(residual connection)和层标准化(layer normalization)。可以形式化表示为:
接着作者使用了一维卷积(one-dimensional CNNs)和最大池化(max pooling)操作来提取高级特征,可以将这个特征映射表示为:最后,则可分别得到药物和靶标蛋白的不同粒度特征:,,,.
3.4 蛋白-配体预测结构亲和力预测
由于不同粒度的子序列和原子级别的特征存在重叠(overlap),如果使用简单的拼接操作可能会造成冗余且影响模型的学习能力。为了高效地整合不同粒度的信息,作者设计了一个简单的带有门控机制(gating mechanism)的highway 前馈网络来控制信息的流动。每个highway层包括一个信息携带门(carry gate, Cr)和一个变换门(transform gate, Tr)。为了保持简洁,作者定义。 最后作者采用均方差(Mean square error, MSE)作为loss函数,即 作者也给出了详细的参数设置情况,如表1所示。表1 参数设置情况
4 数据和结果
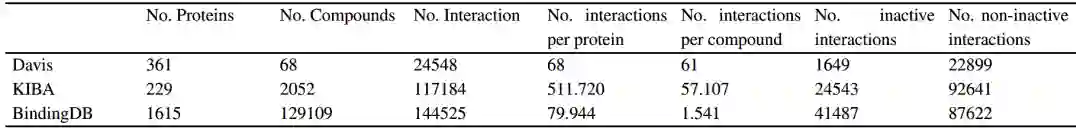
4.1 数据作者采用了3个流行的数据集用于评估模型:KIBA数据集、Davis数据集、Binding DB数据集。数据的统计情况如表2所示。表2 使用的3个数据集的统计情况
4.2 评估指标作者采用了CI、MSE和Pearson相关系数定量衡量模型的性能。
4.3 模型比较
图3 MGPLI和对比方法在Davis、KIBA和BindingDB数据集上的性能表现。误差线表示标准误差。 如图3所示,作者将MGPLI与其他基准模型进行了比较。作者也进行了消减实验、药物的靶标选择性(target selectivity)、PDBBind数据集上的性能评估和比较等分析。
5 总结
在本文中,作者提出了一种新的深度学习模型MGPLI,用于DTA预测任务。MGPLI使用来自药物和蛋白质序列的多粒度信息;也就是说,它整合了SMILES和氨基酸序列中的原子级和子序列级信息。利用这种多粒度信息,MGPLI通过使用transformer编码器和CNN学习原子级和子序列级的表示。为了有效地融合这些表示,MGPLI采用highway层来调节连续训练周期中的信息流。在三个公共数据集上的实验结果表明,MGPLI在随机划分和盲划分场景中都显著优于最先进的模型。尽管MGPLI在PDBbind数据集上的表现并不优于最先进的方法,但本方法仍然是对蛋白质-配体相互作用预测任务上的有效贡献,并可能在未来的工作中进一步增强。
参考资料
Junjie Wang, Jie Hu, Huiting Sun, MengDie Xu, Yun Yu, Yun Liu, Liang Cheng, MGPLI: Exploring Multigranular Representations for Protein-Ligand Interaction Prediction, Bioinformatics, 2022;, btac597,
https://doi.org/10.1093/bioinformatics/btac597
--------- End ---------

