——背景——
现有的基于蛋白结构的深度学习序列设计方法,虽然在测试的计算指标上取得了很好的成果,但是还鲜有方法经过实验的考验仍然超越传统的能量函数方法。基于这一挑战,中国科学技术大学的刘海燕教授课题组,发展了名为ABACUS-R方法,相关工作名为Rotamer-free protein sequence design based on deep learning and self-consistency,于近期发表在Nature Computational Science上。
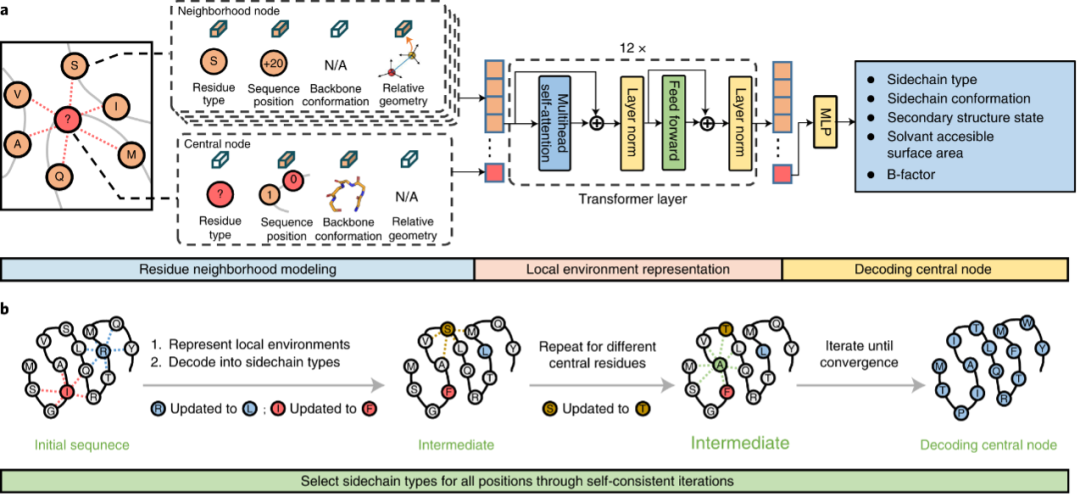
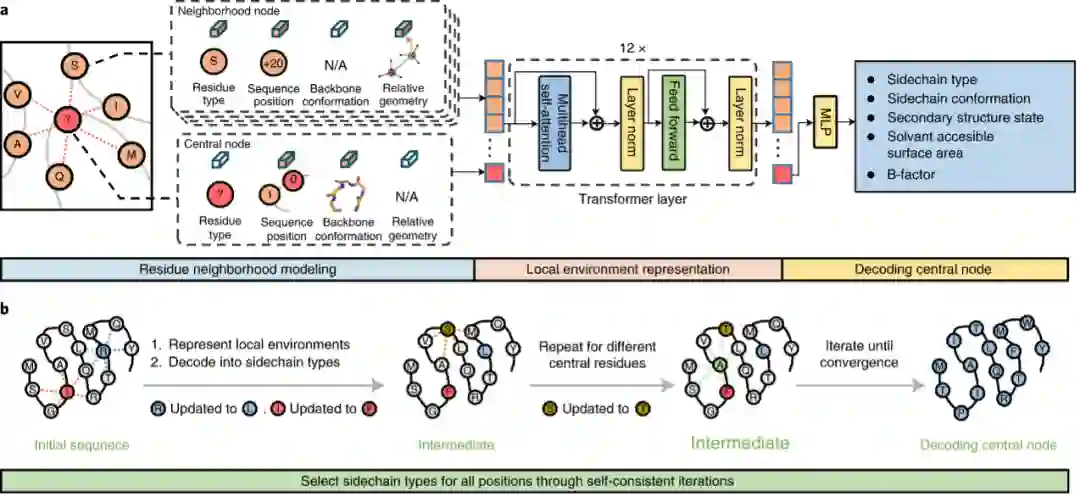
图1. ABACUS-R方法的示意图 ——方法——
ABACUS-R方法包含两部分:(1)一个encoder-decoder网络被预训练用以推断给定骨架的局部环境时中心残基的侧链类型 (2)用该encoder-decoder网络连续更新每个残基的类型,最终收敛获得自洽(self-consistent)。网络的输入是中心残基与空间上最邻近(Cα间距离)k个残基组成的局部结构。邻近残基的特征包含空间层面的相对位置与取向信息(XSPA)、序列层面的相对位置信息(XRSP)以及邻近残基的残基类型(XAA)。第i个中心残基的特征包含全零的XSPA、被mask的XAA以及骨架上的15个ϕi−2, ψi−2, ωi−2 ⋯ ϕi+2, ψi+2, ωi+2,这些特征组合起来会被映射到与邻近残基特征相同的维度。以上模型输入的信息都是旋转平移不变的。局部结构中的所有残基的特征经过可学习的映射后融合后,得到每个残基总特征En。{En; n = 0, 1, 2, ... , k}经过基于transformer架构的encoder-decoder,预测每个中心残基的类型以及其他辅助任务。 自洽迭代设计的方法是:对序列随机初始化,第一轮随机选择80%的残基通过encoder-decoder并行预测其残基类型,以后每轮随机选择的残基数目逐渐下降。最终的设计结果会逐渐收敛。 作者将PDB中的非冗余结构按照两种不同的方式划分了95%作为训练集、5%作为测试集,第一种划分方式确保测试集的结构不会存在训练集中出现过的CATH拓扑,训练得到的模型为Modeleval;第二种划分方式时随机划分Modelfinal。Modeleval可以用来评估模型能力的无偏向性的表现,而Modelfinal使用了更丰富的数据训练表现应当更好。 ——表现评估——
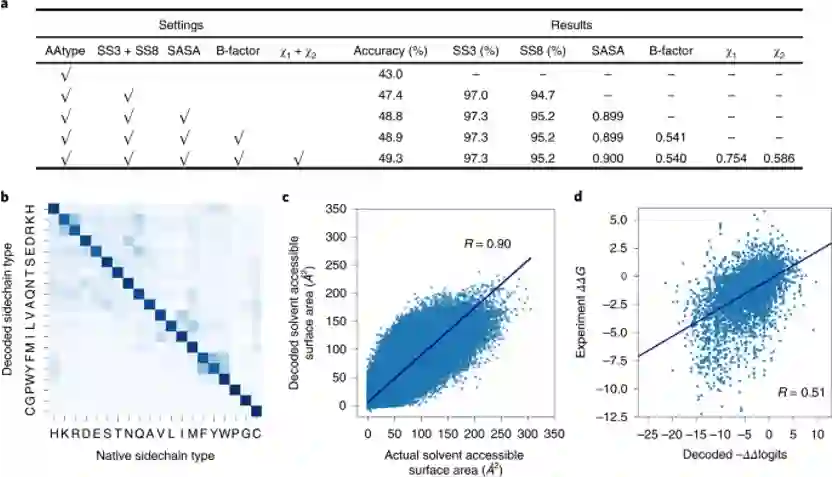
Encoder-decoder的架构可以进行多任务学习,除了训练序列的恢复的任务以外,还可以预测二级结构、SASA、B-factor与侧链扭转角χ1、χ2。多个任务可以增强模型设计序列的能力(图2a),Modeleval与Modelfinal都可以在测试集上最好取得50%左右准确度。在测试集上的结果显示,虽然有些残基类型没有恢复正确,但是模型也学习到了替换为性质相似的残基(图2b)。
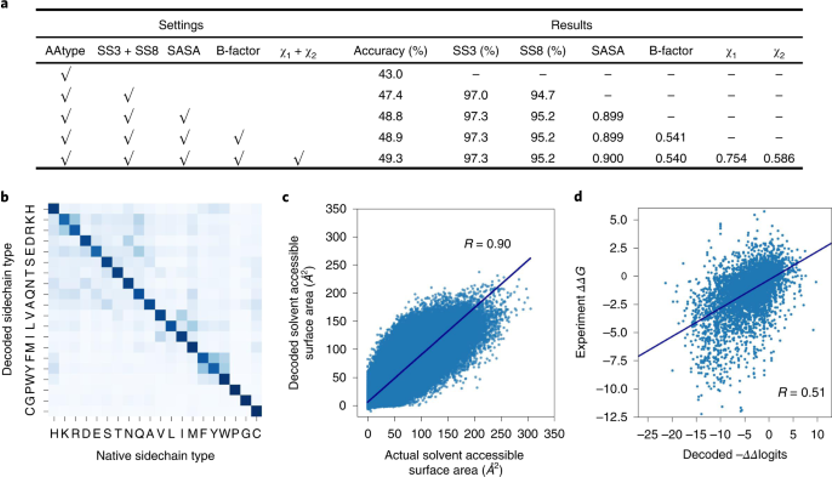
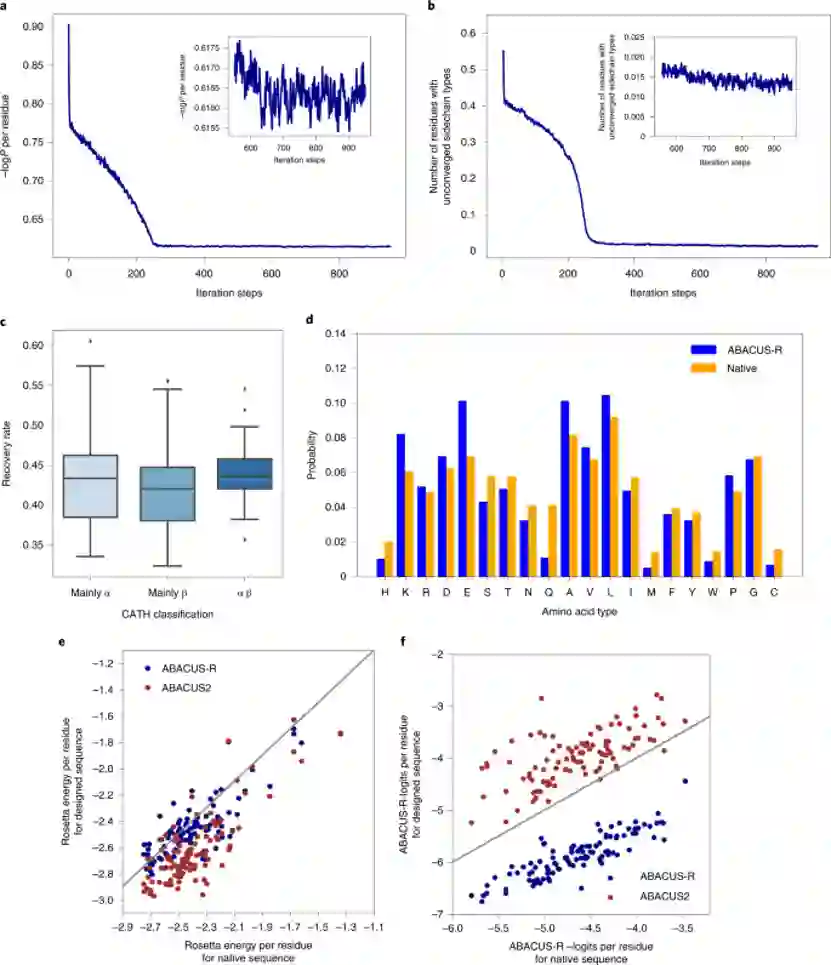
图2. Modeleval在不同任务类型下的表现 Decoder网络输出的是每个位置上残基类型的-logP,类似于选择不同残基对应的能量,所以作者将ProTherm数据集中蛋白突变的ΔΔG与模型计算出相应的−ΔΔlogits进行了比较,发现二者有一定的相关性(图2d),说明模型一定程度上学习到了能量。 接着,作者验证了模型的自洽性,测试集中100个蛋白属于CATH的三个大类,对其中的每个蛋白从随机序列出发设计10条序列,随着迭代的次数变多,平均-logP会趋于收敛(图3a),同时未收敛的残基比例也会收敛(图3b)。不同CATH类别的骨架上取得的序列恢复率差距不大(图3c)。同一蛋白骨架设计出的序列会有很高的相似性(0.76-0.89)。设计出的序列与天然序列相比,序列的成分高度相似(图3d),Pearson相关系数达到了0.93,但GLU、ALA与LYS出现得更频繁,而Gln、His、Met出现得更少。此外,ABACUS-R设计出的序列与ABACUS设计出的序列相比,平均每个残基的Rosetta打分更低(图3e),而平均的-logP打分却更高(图3f),这意味着ABACUS-R学习到的能量与Rosetta打分函数存在正交的部分。
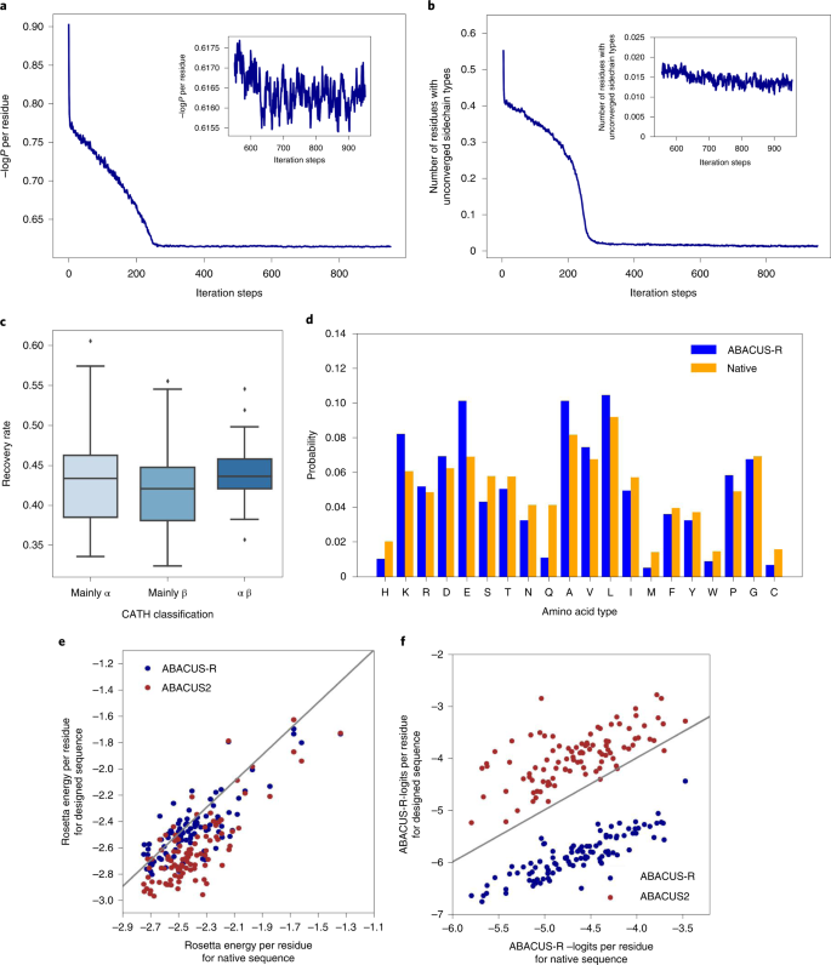
图3. ABACUS-R的自洽能力、设计能力以及学习到的能量与Rosetta打分的比较 相较于其他深度学习方法在单个残基恢复任务上的表现,ABACUS-R超过了除DenseCPD外的所有方法(表1),在整条序列重设计任务上ABACUS-R在两个测试集上都取得了最好的表现(表2)。 表1. ABACUS-R相较其他深度学习方法在单个残基恢复任务上的表现

表2. ABACUS-R相较其他深度学习方法在整条序列重设计任务上的表现

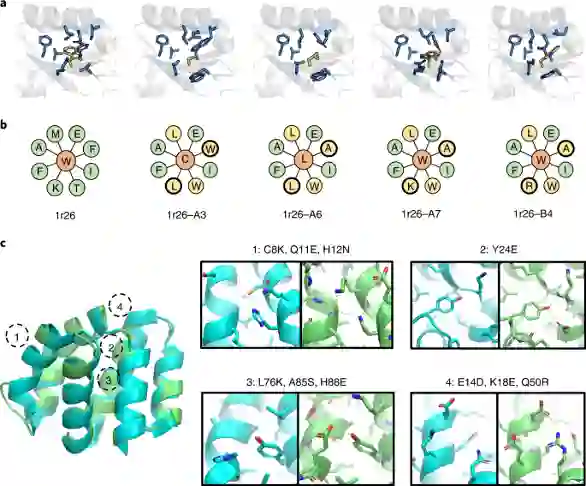
最后,作者在3种天然骨架(PDB ID: 1r26, 1cy5 and 1ubq)上通过实验验证了ABACUS-R的设计能力。设计的方法有两种:第一种采用迭代自洽的设计方法(生成序列的多样性低),第二种采用迭代时对decoder输出结果进行采样(生成序列的多样性高,但-logP能量也略高)。 第一种方法设计的27条序列有26条成功表达,体积排阻色谱与1H NMR实验结果显示所有的蛋白都以单体形式存在,示差扫描量热实验显示5条序列有很好的热稳定性( 97~117 ∘C )。最终,1r26的3个设计与1cy5的1个设计成功解出了晶体结构,Cα RMSD位于0.51~0.88 Å,而1ubq的1个设计虽然没有解出结构,但已有的实验结果显示它折叠成了明确的三维结构。 第二种方法对同一骨架设计的序列相似度在58%左右。30条设计的序列中,25条被成功表达,23条能被可溶地纯化。所有设计同样都是单体存在并且折叠成了明确的三维结构,5个设计有很好的热稳定性(85~118 ∘C)。最终,1r26的1个设计被成功解出了晶体结构,Cα RMSD为0.67 Å。相较方法一的自洽设计,方法二设计成功率下降,成功设计的蛋白热稳定性也略微下降,但作者认为可以接受。 最后,作者展示了所有1r26设计晶体结构核心的侧链pack(图4a,b),以及1cy5设计晶体结构的侧链的极性作用(图4c),说明了ABACUS-R学会了设计侧链的组合以pack好的结构。
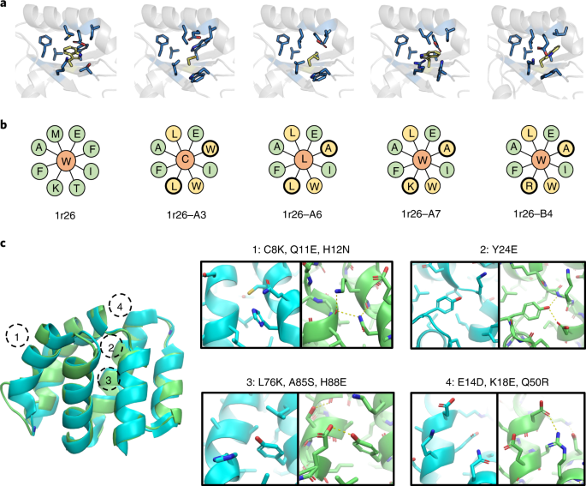
图4. 晶体结构侧链的细节 ——总结——
总之,作者开发的ABACUS-R方法在不需要显示地模拟侧链,可以学习到给定结构下侧链类型的能量打分。ABACUS-R不仅取得了很好的序列恢复度,还在实验上取得了很好的成功率。 参考文献**:**
Liu, Y., et al., Rotamer-free protein sequence design based on deep learning and self-consistency.Nature Computational Science 2022.点击左下角的"阅读原文"即可查看原文章。
作者:顾仲晖审稿:刘佳乐编辑:黄志贤 GoDesignID:Molecular_Design_Lab( 扫描下方二维码可以订阅哦!)