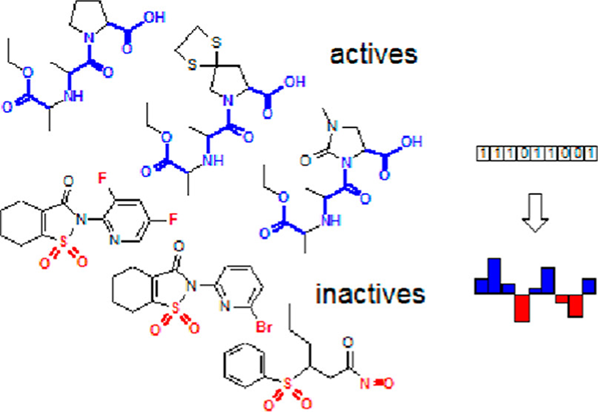
基于配体的虚拟筛选是根据已知活性的小分子,在化合物库中寻找物理或化学特征类似的结构作为候选化合物。这种专注于配体的方法不但速度上具有优势、成功率可观,而且使用也十分便捷,一般只需要用户输入一个代表性的分子结构作为检索模板即可。然而,单一的输入会造成数据的浪费,其他活性或无活性数据也可能蕴藏着重要的信息。针对该问题,来自德国萨尔大学的Michael C. Hutter教授开发了一套累加式分子指纹算法,可以在计算时将所有的结构-活性数据都纳入考量,有效提升了实验数据的利用率。该项研究成果近期发表在美国化学会出版的计算化学和化学信息学核心期刊Journal of Chemical Information and Modeling上【1】。分子指纹与Tanimoto系数是计算机表征分子和比较分子相似度的经典办法。前者种类多样,一般是将化合物转换成比特串,特定位置的“1”与“0”分别表示预设子结构的存在与否,例如一个位置的“1”可以表示磺酰基的存在,“0”则不存在。后者是对两个分子指纹的共同部分进行统计,最终以0到1数字的形式量化表示两个化合物之间的相似性。在此基础上,累加式分子指纹算法则是进行了一定的调整与改良。首先,只选用化合物子结构能与比特位信息一一对应的无压缩型分子指纹(MACCS与PubChem)来表征分子。累加过程中,不同子结构出现在活性分子中的累计次数会除以活性分子总数,计算结果以正数的形式记录在对应比特位,无活性化合物也进行同样的分析,但以负数的形式体现**(图-1)**。然后,因为数学原因(分母不能为0),以Sørensen−Dice系数取代Tanimoto系数进行相似性比较,取值范围-1到1,正数结果表示检索化合物预期是有活性的,负数则相反。除了以0为边界的正负数分类外,研究者还补充了贝叶斯分类算法作为判断化合物有/无活性的可选方法。
图1, 累加式分子指纹算法对活性/无活性化合物高频子结构的统计示意为了验证有效性,研究者考察了算法在血管紧张素转换酶ACE等多个靶点虚筛场景中的表现。首先在有效数据的利用上,发现算法确实能总结出代表性的子结构信息,例如在ACE靶点的训练集中,算法解析到羧酸、C-O、叔胺等是高频活性子结构**(图-2)**,有利于活性,这些特征也可以在上市的ACE抑制剂中观察到。而N-O、卤元素等则不利于活性,被发现是无活性化合物的常见片段。虚筛性能方面,以0为活性区分边界的算法可以找回所有的ACE靶点真实活性分子,灵敏度为1,特异性、准确性和AUC分别是0.413/0.593/0.962;以贝叶斯分类作为活性判断依据的算法,灵敏度、特异性、准确性和AUC分别是0.997/0.900/0.947/0.995。相比之下,后者对无活性或诱饵(Decoy)分子的识别有显著改善,但在另一项类似富集率的评测指标BEDROC上,前者又显示能更早的找回活性分子,表明这两种方法是各有优势的。
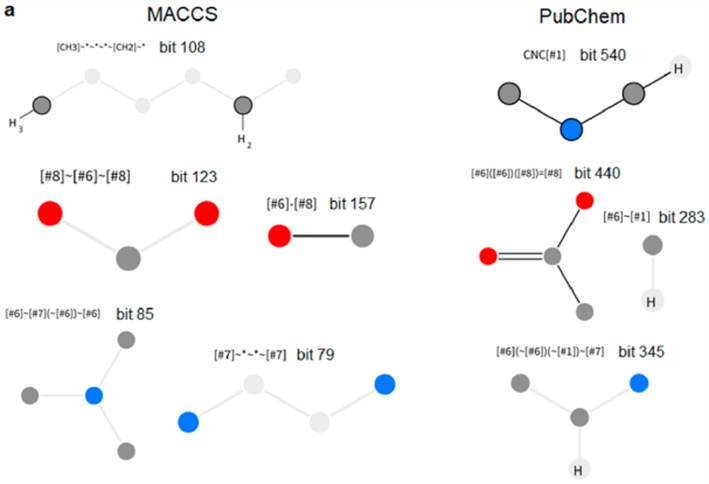
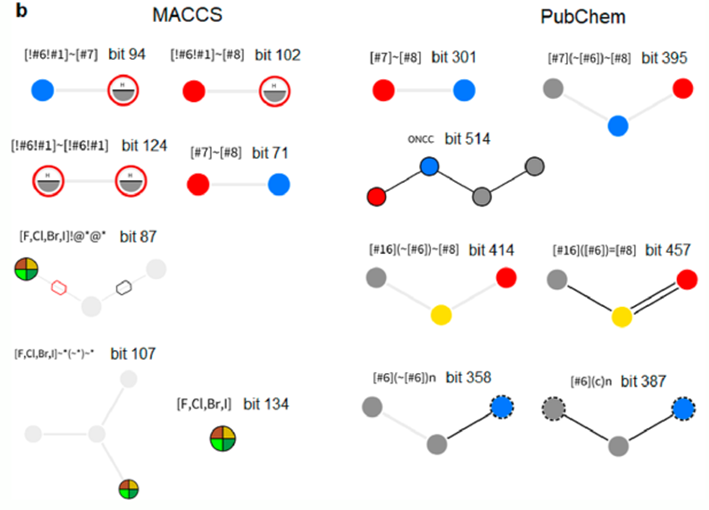
图-2, 累加式分子指纹算法记录的ACE靶点高频活性/无活性子结构信息。a)左为利用MACCS表征化合物时算法统计的高频活性子结构,右为PubChem结果;b)无活性分子高频子结构信息。虚线圆圈表示芳环,不同颜色表示不同元素,红圈表示禁止该原子,淡灰色表示任意原子或连接键,六边形表示环结构,右上角比特位表示该子结构在预设比特串中的位置。【小结】在传统基于配体的虚拟筛选方法上,研究者设计了一套新颖的累加式分子指纹算法,实现了分子指纹与实验数据的有机结合,继承前者速度优势的同时,信息利用率更高,虚拟筛选实战表现优异,可作为药物发现的又一项便捷工具。参考文献【1】Hutter M C. Differential Multimolecule Fingerprint for Similarity Search─ Making Use of Active and Inactive Compound Sets in Virtual Screening. Journal of Chemical Information and Modeling, 2022, 62(11): 2726–2736. doi.10.1021/acs.jcim.2c00242