这篇文献,是近两年DDI(Drug-Drug interactions)预测的相关文献中,引用相对较高的文献。
你可能已经看到,有些DDI类预测论文会把本篇论文的结果,作为基线进行比较。比如在今年7月份发表的深度学习多模态的DDI预测[1]论文,还有比较火的Graph embedding在DDI上的应用等等。
![]()
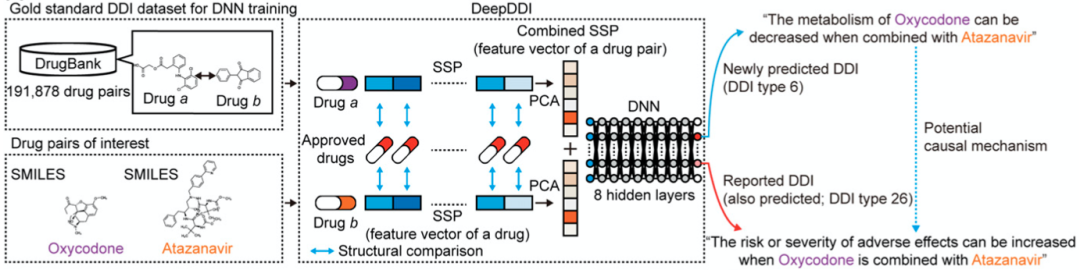
仅需Drug name和Drug structural information作为输入,就可以做四类任务:1)预测引起ADEs(adverse drug events)的DDI;2)可选择的药物推荐;3)预测食物成分和药物交互的影响;4) 食物成分的活性预测。
![]()
DeepDDI由两部分组成:a. 结构相似谱(Structural similarity profile)SSP; b.深度神经网络。
其实SSP就是通过计算药物和药物化学结构的相似性后得到的一个向量表征。具体在下一个小节特征表征中解释。
DNN是一个多标签的模型,有8个隐藏层的简单神经网络。DeepDDI网络输入:a. SMILES格式的化学结构; b. 一对药物的名字。
DeepDDI输出是预测的86个DDI types概率。如果某一类的概率大于阈值0.47,就认为出现这个DDI type。
DeepDDI的训练数据是来自DrugBank中覆盖了191878 药物对的标准DDI数据集。
SSP(Structural similarity profile)
![]()
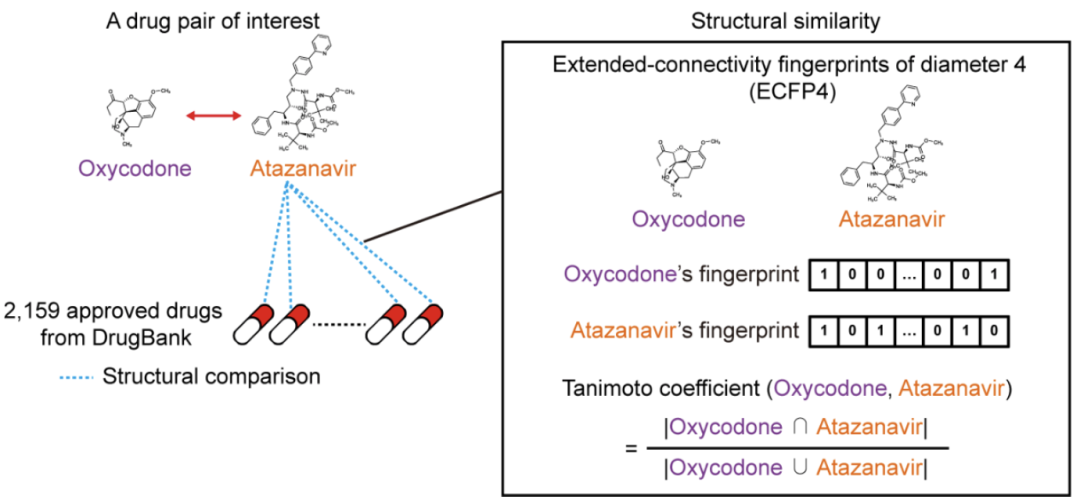
模型的输入是两个药物化学结构的smile格式,两个药物中的每个药物会和DrugBank中的2159个每个药物计算相似性,组成2159维的向量。
一个药物和一个药物计算Structural similarity方法如上图的黑色框。
输入为两个药物的化学结构smile格式,计算药物与药物的化学结构相似性。首先需要考虑,怎么表征化学分子,最后考虑用什么度量化学结构与化学结构的相似性。
通用的做法: 分子片段用比特向量(比特位)表示,即0和1值,然后用Tanimoto coefficient(也叫Jaccard系数)计算相似性。
分子指纹上的每个比特位对应于一种分子片段,假设相似的分子之间必然有许多公共的片段,那么具有相似指纹的分子具有很大的概率在2D结构上也是相似的。
有十多种方法可以评估两个向量之间的相似性(Fingerprints-Screening and Similarity),最常见的是欧几里德距离。
但是对于分子指纹,行业标准是Tanimoto系数,它由两个指纹中设置为1的公共位数除以两个指纹之间设置为1的总位数组成。
![]()
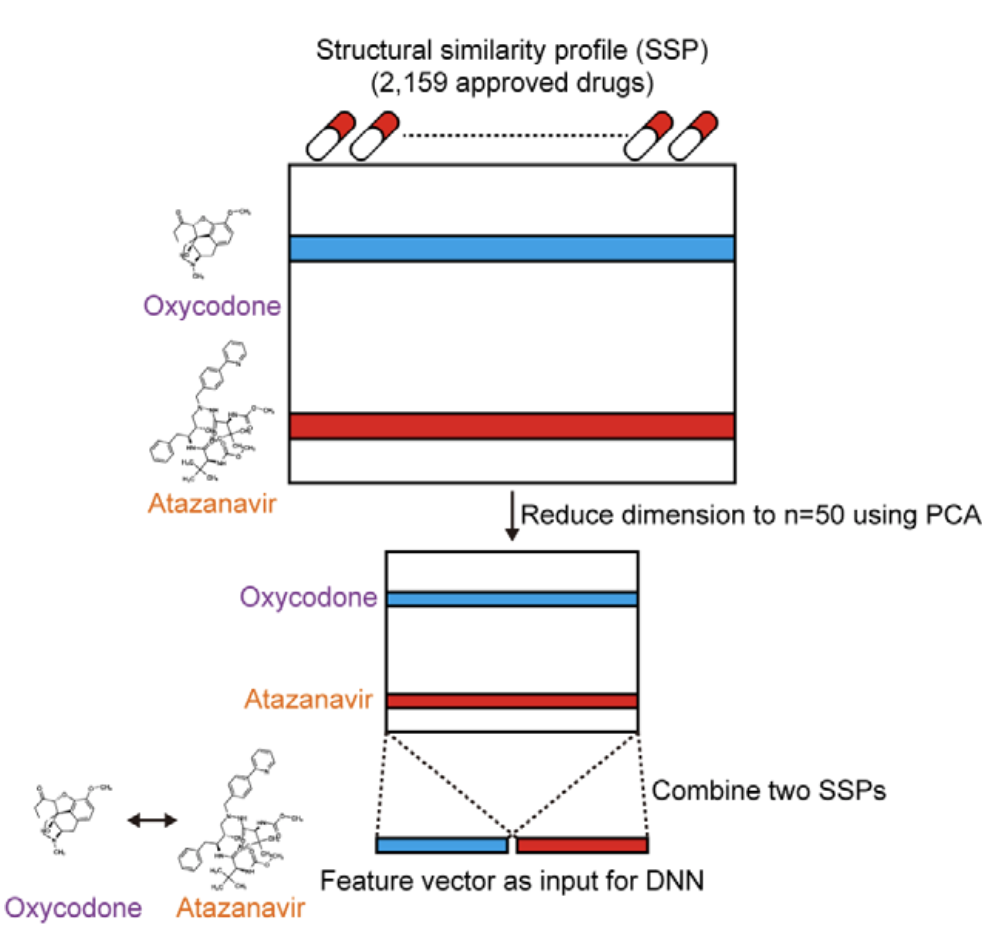
得到SSP,它是对药物表示的2159维特征向量。把两个输入药物的特征向量,都用PCA降到50维的向量,然后将两个药物的特征表示concat一起,得到100维的Feature vector作为DNN的输入。
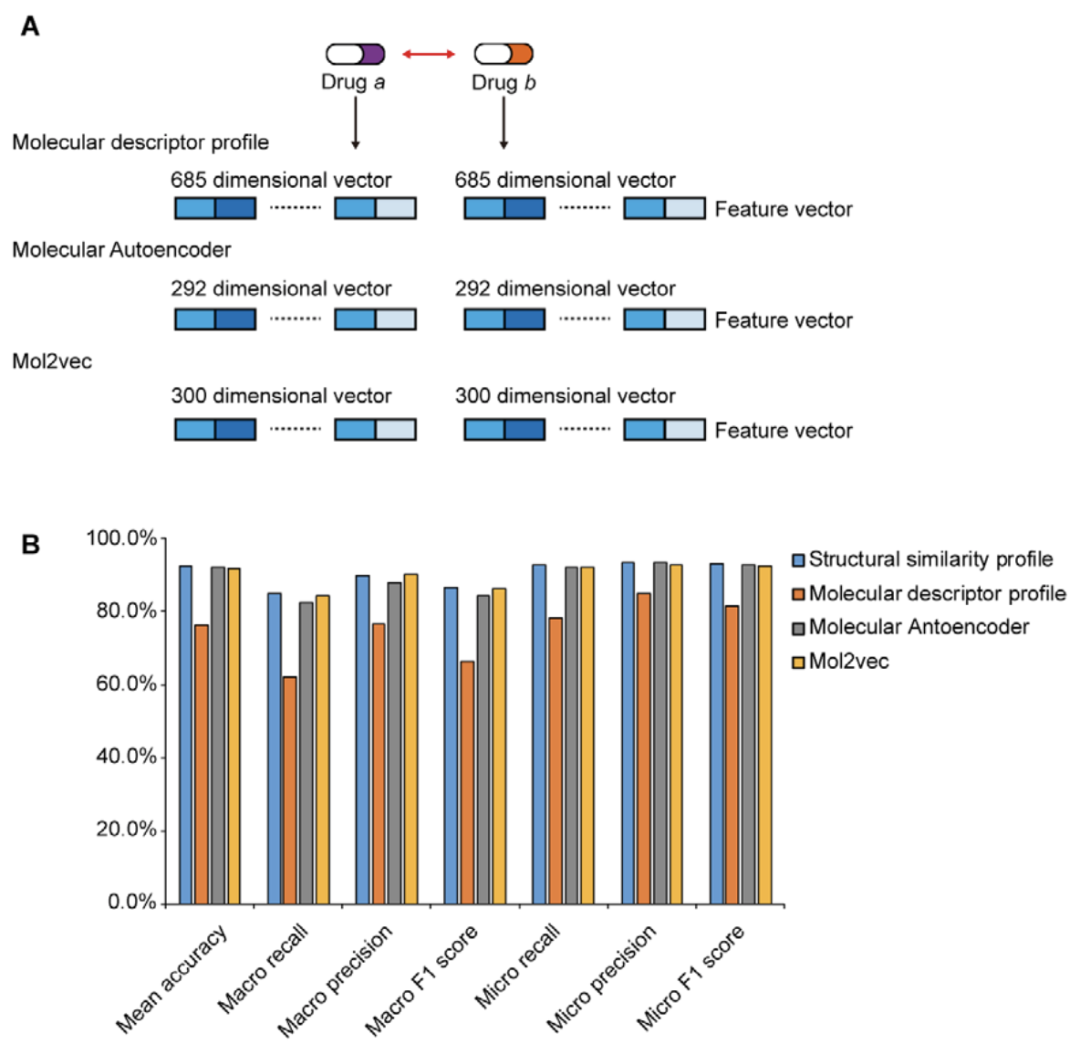
作者除了考虑SSP这种特征表示,还考虑了Molecular descriptor profile、Molecular Autoencoder和Mol2vec这三种特征表示(如下图A所示),发现SSP比Molecular Autoencoder和Mol2vec略微有点优势(如下图B所示)。
![]()
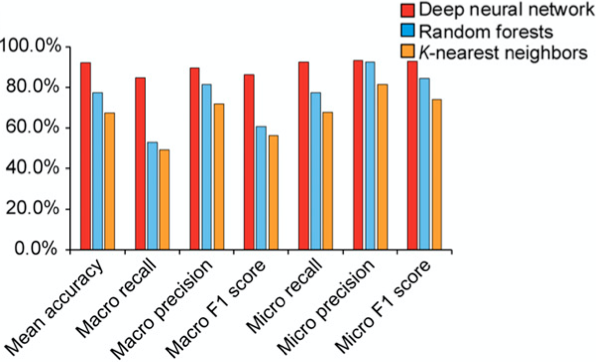
作者也同随机森林和K近邻算法进行比较,发现深度神经网络比其它两个模型更好,如下图。
![]()
如果能增加DDI的数据量,预测准确率还会有提升;考虑到药物和代谢物的浓度以及两个给定分子对其靶点的体内亲和力对DDIs和DFIs的影响,未来还需开发算法。
DeepDDI将成为分析药物或食物成分对的必要工具,并可进一步扩展到未来对多种化合物的“真实世界”DDI和DFI研究。
https://bitbucket.org/kaistsystemsbiology/deepddi