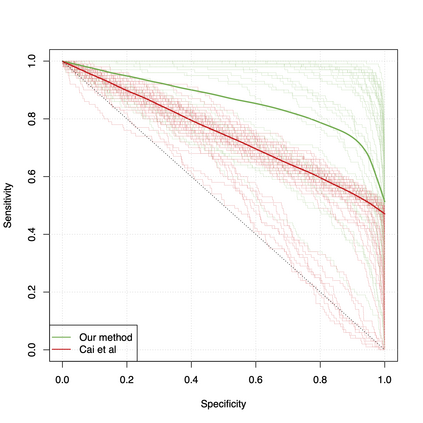
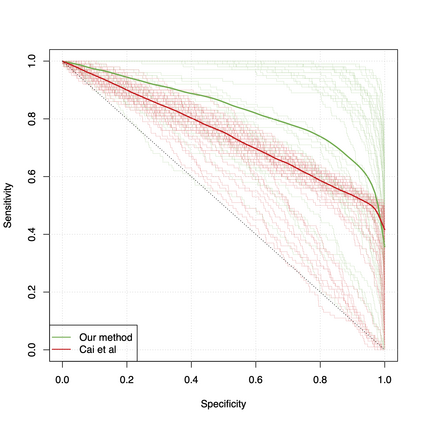
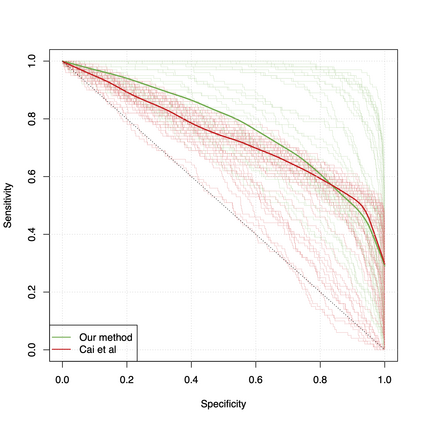
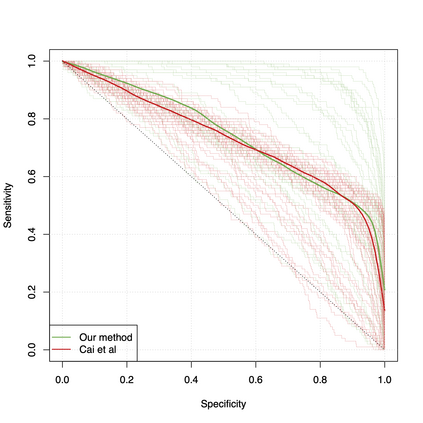
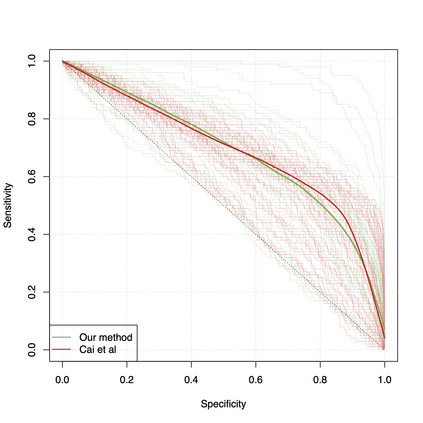
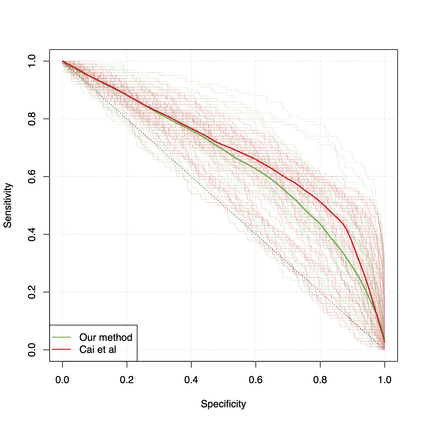
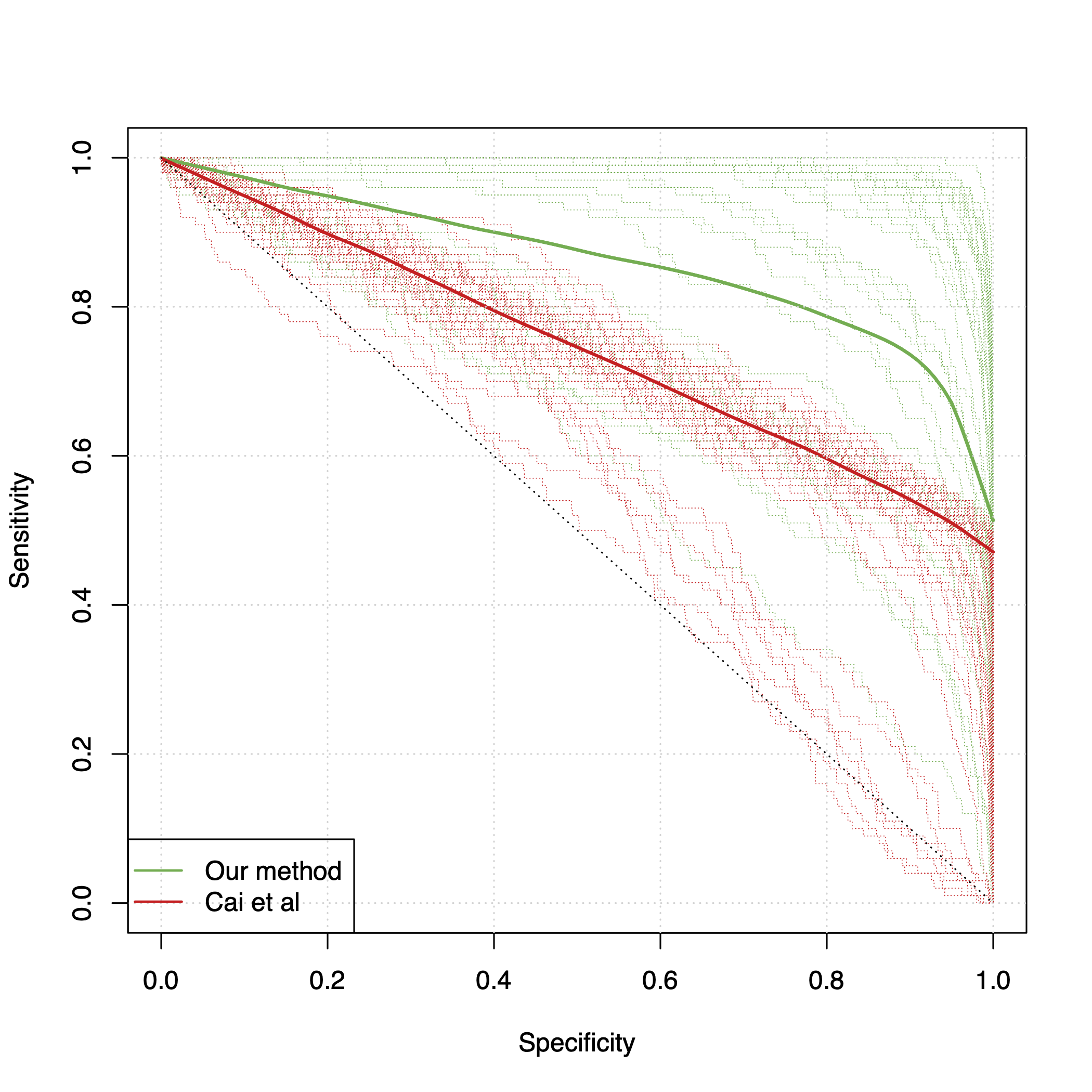
Differential co-expression analysis has been widely applied by scientists in understanding the biological mechanisms of diseases. However, the unknown differential patterns are often complicated; thus, models based on simplified parametric assumptions can be ineffective in identifying the differences. Meanwhile, the gene expression data involved in such analysis are in extremely high dimensions by nature, whose correlation matrices may not even be computable. Such a large scale seriously limits the application of most well-studied statistical methods. This paper introduces a simple yet powerful approach to the differential correlation analysis problem called compressed spectral screening. By leveraging spectral structures and random sampling techniques, our approach could achieve a highly accurate screening of features with complicated differential patterns while maintaining the scalability to analyze correlation matrices of $10^4$--$10^5$ variables within a few minutes on a standard personal computer. We have applied this screening approach in comparing a TCGA data set about Glioblastoma with normal subjects. Our analysis successfully identifies multiple functional modules of genes that exhibit different co-expression patterns. The findings reveal new insights about Glioblastoma's evolving mechanism. The validity of our approach is also justified by a theoretical analysis, showing that the compressed spectral analysis can achieve variable screening consistency.
翻译:科学家在了解疾病生物机制时广泛应用了差异共表达分析,但科学家们在理解疾病的生物机制时广泛应用了差异共表达分析,但未知的差异模式往往很复杂;因此,基于简化参数假设的模型在识别差异方面可能无效;同时,这种分析所涉及的基因表达数据自然具有极高的层面,其关联矩阵可能甚至无法计算。如此大规模地严重限制了最经过广泛研究的统计方法的应用。本文对差异相关分析问题采用了简单而有力的方法,称为压缩光谱筛选。通过利用光谱结构和随机抽样技术,我们的方法可以非常准确地筛选具有复杂差异模式的特征,同时在标准的个人计算机上维持在几分钟内分析10美元-10美元-5美元相关变量的可缩缩缩性。我们采用了这种筛选方法,将关于Glioblastoma的TCGA数据集与正常主题进行比较。我们的分析成功地确定了显示不同共表形态的基因的多个功能模块。通过利用光谱结构和随机抽样技术,发现关于Gliobastoma的演变机制的新见解。我们的方法的有效性也通过理论分析得到证明。