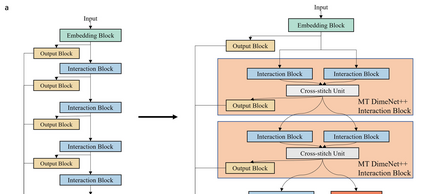
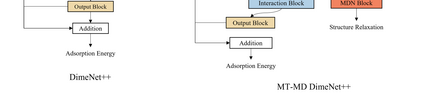
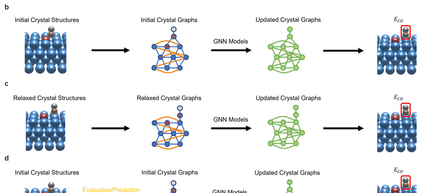
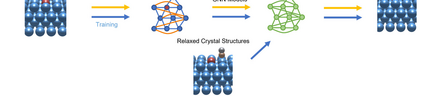
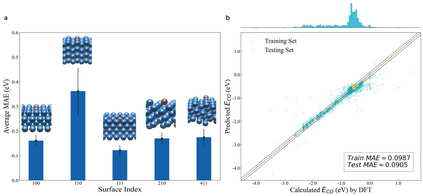
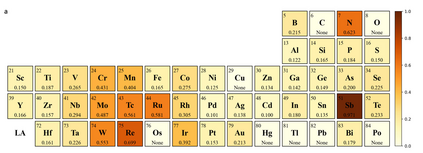
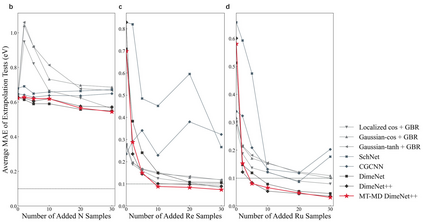
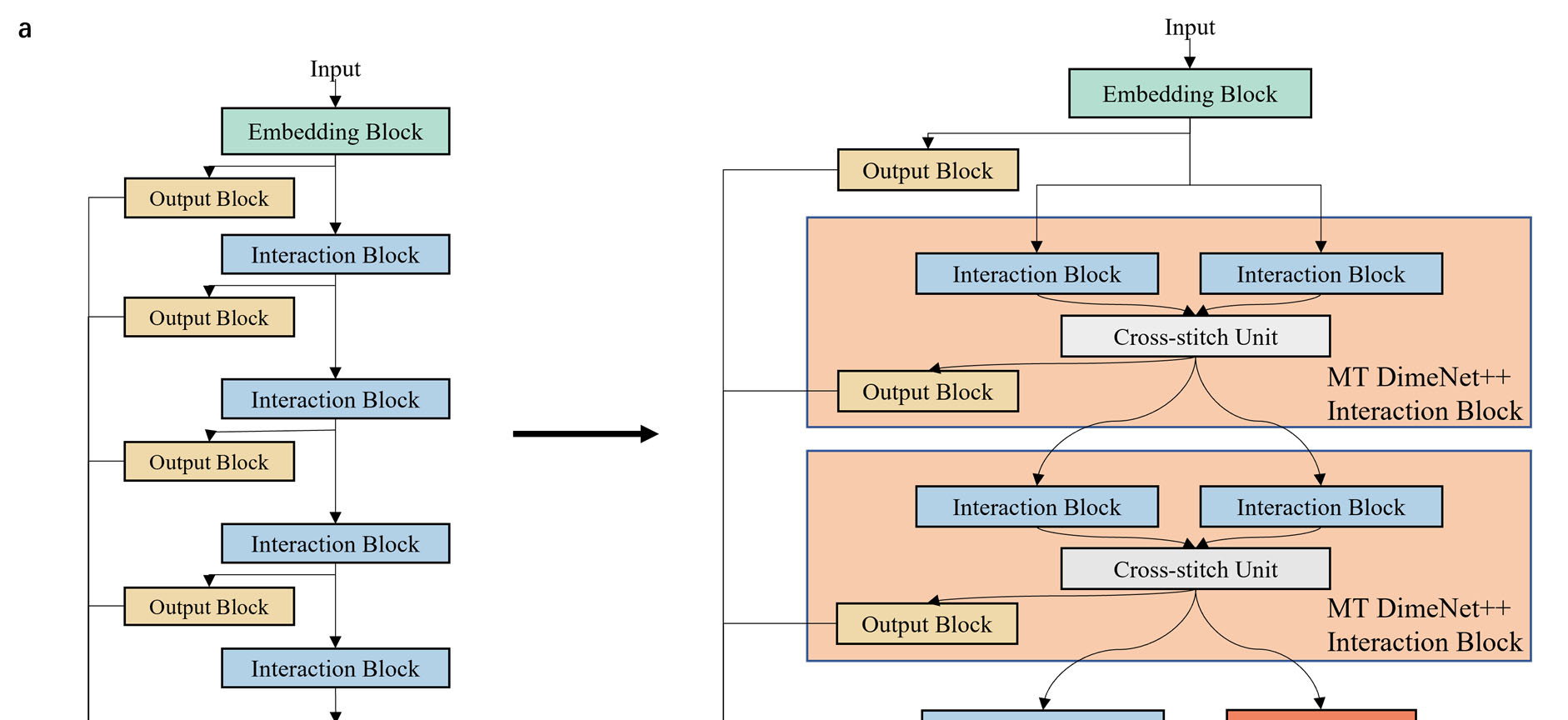
Graph neural networks (GNNs) have drawn more and more attention from material scientists and demonstrated a high capacity to establish connections between the structure and properties. However, with only unrelaxed structures provided as input, few GNN models can predict the thermodynamic properties of relaxed configurations with an acceptable level of error. In this work, we develop a multi-task (MT) architecture based on DimeNet++ and mixture density networks to improve the performance of such task. Taking CO adsorption on Cu-based single-atom alloy catalysts as an illustration, we show that our method can reliably estimate CO adsorption energy with a mean absolute error of 0.087 eV from the initial CO adsorption structures without costly first-principles calculations. Further, compared to other state-of-the-art GNN methods, our model exhibits improved generalization ability when predicting catalytic performance of out-of-domain configurations, built with either unseen substrate surfaces or doping species. We show that the proposed MT GNN strategy can facilitate catalyst discovery.
翻译:脑电图网络(GNNs)吸引了物质科学家越来越多的关注,并展示了在结构和特性之间建立联系的高度能力;然而,由于作为投入而提供的只是不松动结构,很少有GNN模型能够预测松动配置的热力特性,并有可接受的误差;在这项工作中,我们根据DimeNet+++和混合物密度网络开发了一个多任务(MT)结构,以改进这项任务的绩效;用CO对基于Cu的单原子合金催化剂的吸附作为示例,我们表明,我们的方法可以可靠地估计CO吸收能量,从最初的CO吸附结构中得出0.087 eV的绝对误差,而没有代价高昂的第一条原则计算;此外,与其他最先进的GNNN方法相比,我们模型展示了在预测以未知的子表层表面或剂量物种建造的外部配置的催化性能时,提高了一般化能力。我们表明,拟议的MT GNNN战略可以促进催化剂的发现。