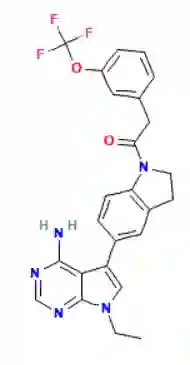
Identifying novel drug-target interactions (DTI) is a critical and rate limiting step in drug discovery. While deep learning models have been proposed to accelerate the identification process, we show that state-of-the-art models fail to generalize to novel (i.e., never-before-seen) structures. We first unveil the mechanisms responsible for this shortcoming, demonstrating how models rely on shortcuts that leverage the topology of the protein-ligand bipartite network, rather than learning the node features. Then, we introduce AI-Bind, a pipeline that combines network-based sampling strategies with unsupervised pre-training, allowing us to limit the annotation imbalance and improve binding predictions for novel proteins and ligands. We illustrate the value of AI-Bind by predicting drugs and natural compounds with binding affinity to SARS-CoV-2 viral proteins and the associated human proteins. We also validate these predictions via auto-docking simulations and comparison with recent experimental evidence, and step up the process of interpreting machine learning prediction of protein-ligand binding by identifying potential active binding sites on the amino acid sequence. Overall, AI-Bind offers a powerful high-throughput approach to identify drug-target combinations, with the potential of becoming a powerful tool in drug discovery.
翻译:确定新的药物目标互动(DTI)是药物发现中一个关键且节点有限的步骤。虽然已经提出深层次的学习模式以加快鉴定过程,但我们表明,最先进的模型未能向新颖(即从不见得)结构(即从不见)结构推广。我们首先公布应对这一缺陷负责的机制,展示模型如何依赖利用蛋白质和双边网络的地形学的捷径,而不是学习节点特征。然后,我们引入AI-Bind,这是一个基于网络的采样战略与未经监督的预培训相结合的管道,使我们能够限制注解不平衡,改进对新蛋白和皮层的具有约束力的预测。我们通过预测与SAS-COV-2病毒蛋白和相关人类蛋白质相关的结合的药物和自然化合物来说明AI-Bind的价值。我们还通过自动摄取模拟和与最近实验证据的比较来验证这些预测,并通过确定具有潜在约束力的、具有强大影响力的药物稳定度预测方法来解释蛋白和蛋白质的机学预测过程,从而确定具有强大的药效性、具有潜在坚固度的实验室级的实验室。