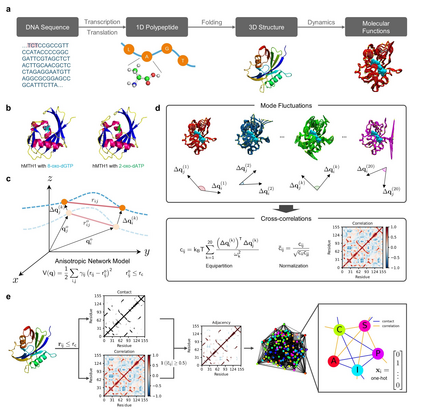
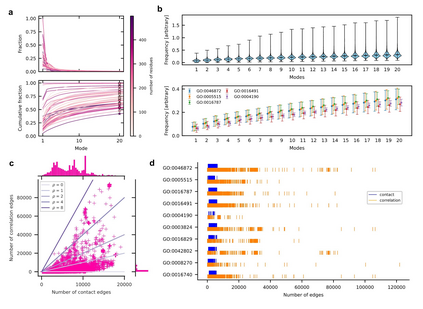
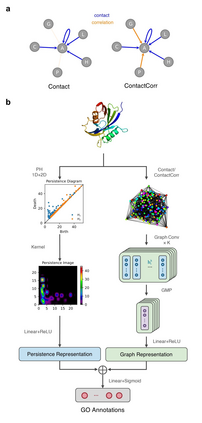
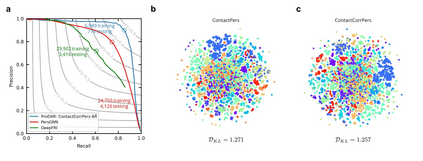
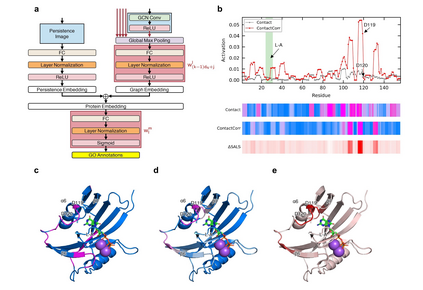
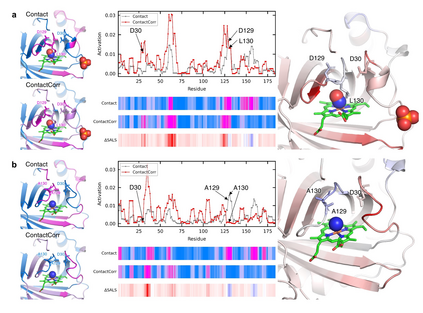
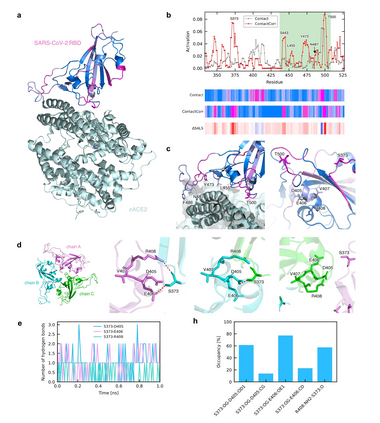
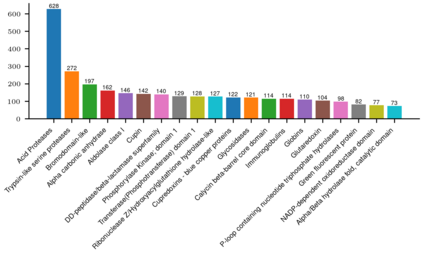
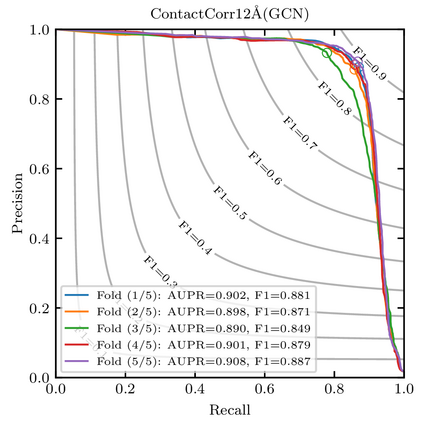
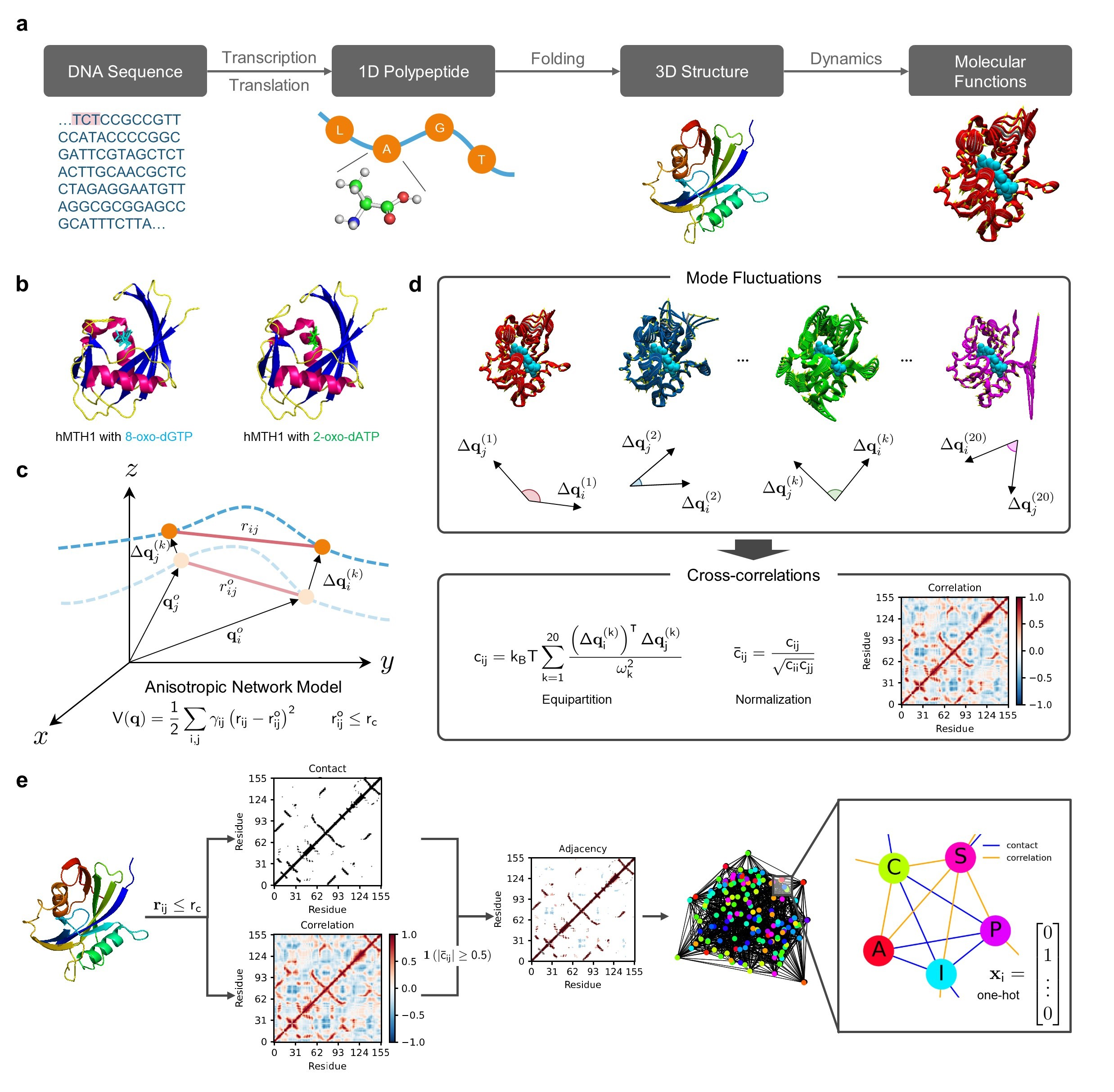
Recent advances in protein function prediction exploit graph-based deep learning approaches to correlate the structural and topological features of proteins with their molecular functions. However, proteins in vivo are not static but dynamic molecules that alter conformation for functional purposes. Here we apply normal mode analysis to native protein conformations and augment protein graphs by connecting edges between dynamically correlated residue pairs. In the multilabel function classification task, our method demonstrates a remarkable performance gain based on this dynamics-informed representation. The proposed graph neural network, ProDAR, increases the interpretability and generalizability of residue-level annotations and robustly reflects structural nuance in proteins. We elucidate the importance of dynamic information in graph representation by comparing class activation maps for hMTH1, nitrophorin, and SARS-CoV-2 receptor binding domain. Our model successfully learns the dynamic fingerprints of proteins and pinpoints the residues of functional impacts, with vast untapped potential for broad biotechnology and pharmaceutical applications.
翻译:蛋白质功能预测最近的进展利用基于图表的深层学习方法,将蛋白质的结构和地形特征与其分子功能联系起来。然而,活体中的蛋白质不是静态的,而是动态分子,它们改变功能性的一致性。我们在这里对本地蛋白分解进行正常模式分析,并通过将动态相关残余体的边缘连接在一起来增加蛋白质图。在多标签功能分类任务中,我们的方法显示了基于这种动态知情的表达方式而取得的巨大绩效收益。拟议的图表神经网络ProDAR,增加了残留水平说明的可解释性和可概括性,并有力地反映了蛋白质的结构细微。我们通过比较HMTH1、硝营养素和SAS-COV-2受体结合域的类活动图,在图表中说明了动态信息的重要性。我们的模型成功地学习了蛋白质的动态指纹,并确定了功能影响残余物,为广泛的生物技术和药物应用提供了巨大的未开发潜力。