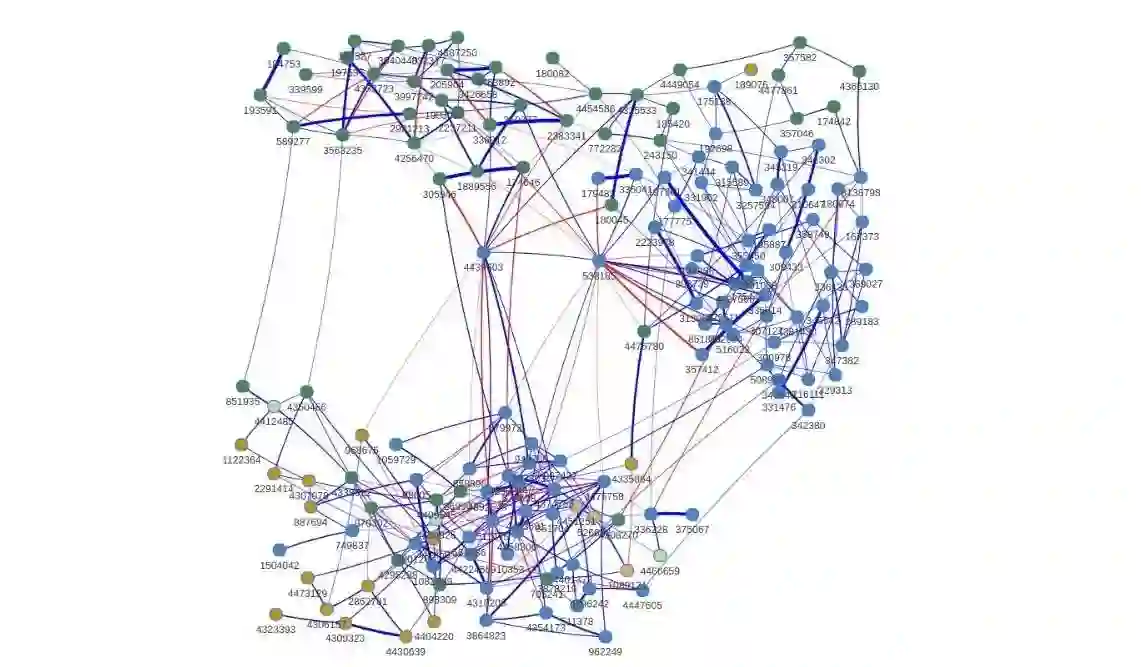
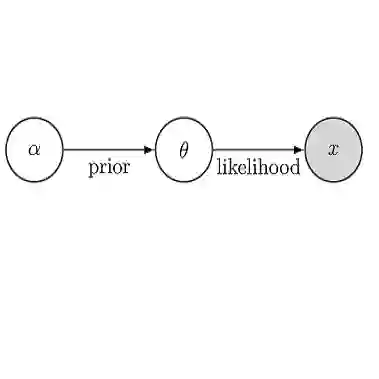
Metagenomics combined with high-resolution sequencing have enabled researchers to study the genomes of entire microbial communities. Revealing the underlying interactions between these communities is of vital importance to learn how microbes influence human health. Learning these interactions from microbiome data is challenging, due to the high dimensionality, discreteness, broad dispersion levels, compositionality and excess of zero counts that characterize these data. To tackle these issues, we develop a novel Gaussian copula graphical model with two key elements. Firstly, we model the marginal distributions via discrete Weibull regression, both to account for the typical features of microbiome data and to include the dependency from external covariates, often available in genomic studies but rarely used for network inference. Secondly, we advance a Bayesian structure learning framework, based on a computationally efficient search algorithm that is suited to high dimensionality. The approach returns simultaneous inference of the marginal effects and of the dependency structure, including graph uncertainty estimates. A simulation study and a real data analysis of microbiome data highlight the applicability of the proposed approach at inferring networks from high-dimensional count data in general, and its relevance to microbiota data analyses in particular. The proposed method is implemented in the R package BDgraph.
翻译:与高分辨率测序相结合的元基因组使研究人员能够研究整个微生物群落的基因组。揭示这些群落之间的内在互动关系对于了解微生物如何影响人类健康至关重要。从微生物数据中学习这些互动关系具有挑战性,因为这些数据具有高度的维度、离散性、广泛分散程度、构成性和零计过量的特点。为了解决这些问题,我们开发了一个具有两个关键要素的新型高斯氏干毛线图形模型。首先,我们通过离散 Weibull回归模型进行边际分布。我们通过离散 Weibull回归模型进行边际分布,既说明微生物数据典型特征,又包括外部变异器的依赖性,通常在基因学研究中提供,但很少用于网络推断。第二,我们根据适合高维度数据特点的计算高效搜索算法,推出一个巴耶斯结构学习框架。我们的方法是同时推断边际效应和依赖结构的推论,包括图形不确定性估计。对微生物数据进行模拟研究和实际数据分析,强调拟议中的网络推算方法在高度的微生物中具有相关性。在高度分析中采用一般数据和测算方法。