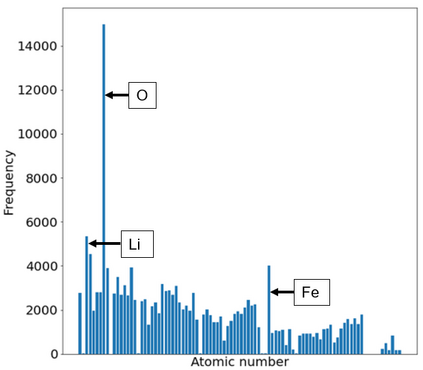
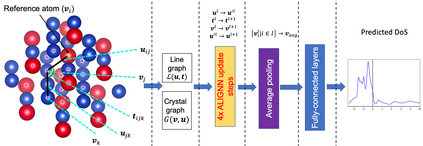
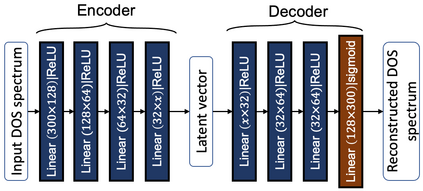
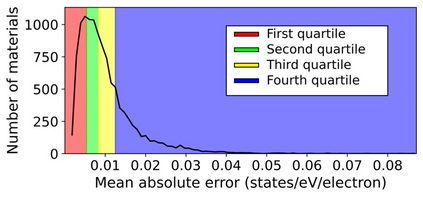
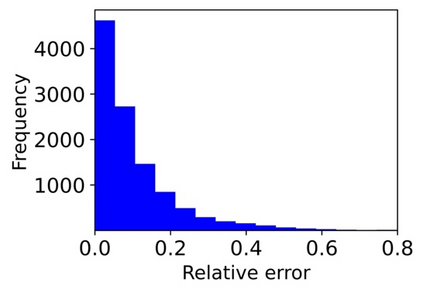
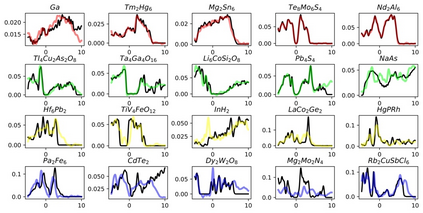
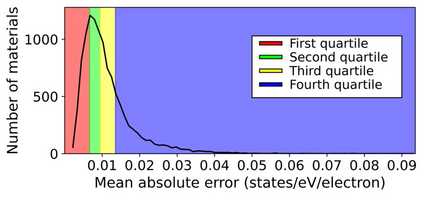
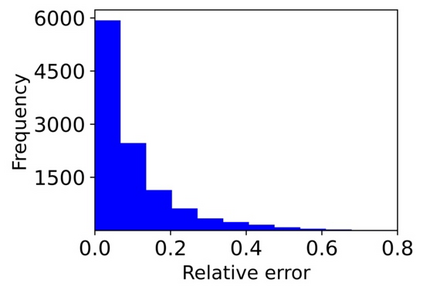
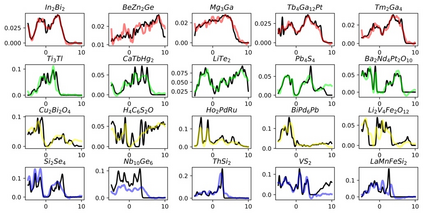
Machine learning (ML) based models have greatly enhanced the traditional materials discovery and design pipeline. Specifically, in recent years, surrogate ML models for material property prediction have demonstrated success in predicting discrete scalar-valued target properties to within reasonable accuracy of their DFT-computed values. However, accurate prediction of spectral targets such as the electron Density of States (DOS) poses a much more challenging problem due to the complexity of the target, and the limited amount of available training data. In this study, we present an extension of the recently developed Atomistic Line Graph Neural Network (ALIGNN) to accurately predict DOS of a large set of material unit cell structures, trained to the publicly available JARVIS-DFT dataset. Furthermore, we evaluate two methods of representation of the target quantity - a direct discretized spectrum, and a compressed low-dimensional representation obtained using an autoencoder. Through this work, we demonstrate the utility of graph-based featurization and modeling methods in the prediction of complex targets that depend on both chemistry and directional characteristics of material structures.
翻译:机器学习模型大大加强了传统材料发现和设计管道,具体来说,近年来,材料财产预测的替代ML模型表明,成功地在其DFT计算值的合理准确性范围内预测离散的卡路里值目标特性,但是,由于目标的复杂性和现有培训数据有限,准确预测国家电子密度等光谱目标带来了更具有挑战性的问题。在本研究中,我们介绍了最近开发的Atomistic Liine 图形神经网络(ALIGN)的扩展,以准确预测DOS的一组大型材料单元细胞结构,这些结构经过培训,可以公开获得JARVIS-DFT数据集。此外,我们评估了两种目标数量的表述方法----一种直接离散频谱,以及一种使用自动编码器获得的压缩低维代表。通过这项工作,我们展示了基于图形的成型成型和建模方法在预测依赖材料结构的化学和定向特性的复杂目标方面的效用。