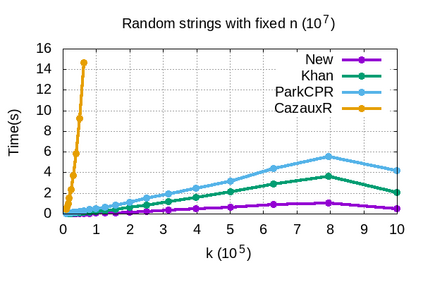
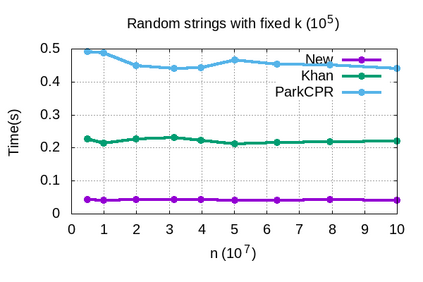
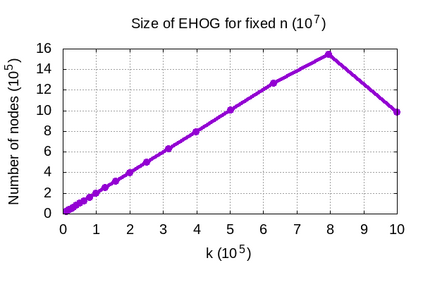
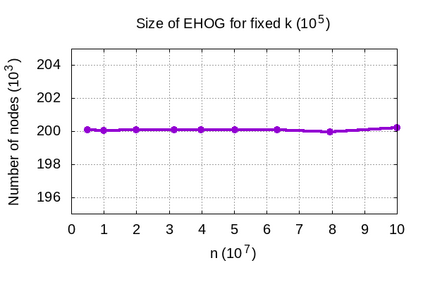
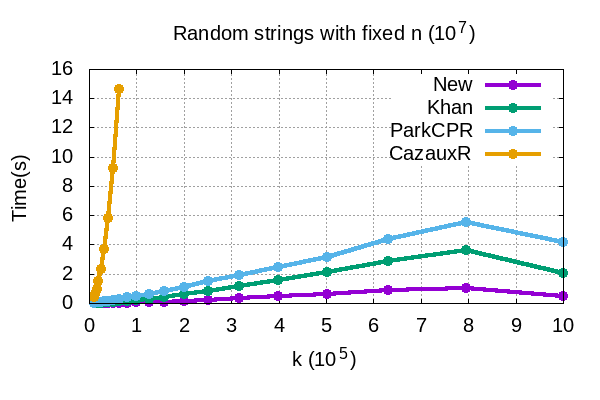
Genome assembly is a prominent problem studied in bioinformatics, which computes the source string using a set of its overlapping substrings. Classically, genome assembly uses assembly graphs built using this set of substrings to compute the source string efficiently, having a tradeoff between scalability and avoiding information loss. The scalable de Bruijn graphs come at the price of losing crucial overlap information. The complete overlap information is stored in overlap graphs using quadratic space. Hierarchical overlap graphs [IPL20] (HOG) overcome these limitations, avoiding information loss despite using linear space. After a series of suboptimal improvements, Khan and Park et al. simultaneously presented two optimal algorithms [CPM2021], where only the former was seemingly practical. We empirically analyze all the practical algorithms for computing HOG, where the optimal algorithm [CPM2021] outperforms the previous algorithms as expected, though at the expense of extra memory. However, it uses non-intuitive approach and non-trivial data structures. We present arguably the most intuitive algorithm, using only elementary arrays, which is also optimal. Our algorithm empirically proves even better for both time and memory over all the algorithms, highlighting its significance in both theory and practice. We further explore the applications of hierarchical overlap graphs to solve various forms of suffix-prefix queries on a set of strings. Loukides et al. [CPM2023] recently presented state-of-the-art algorithms for these queries. However, these algorithms require complex black-box data structures and are seemingly impractical. Our algorithms, despite failing to match the state-of-the-art algorithms theoretically, answer different queries ranging from 0.01-100 milliseconds for a data set having around a billion characters.
翻译:暂无翻译