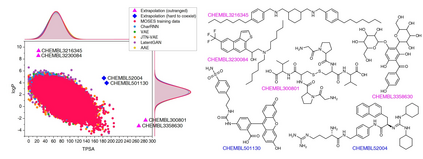
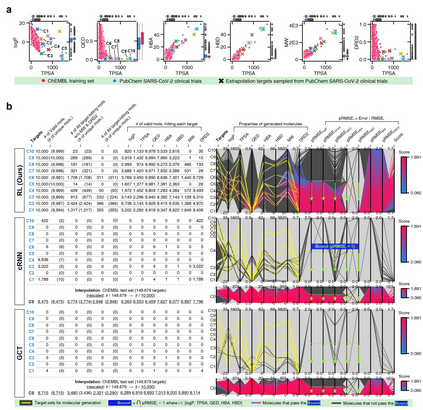
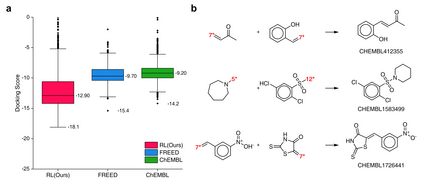
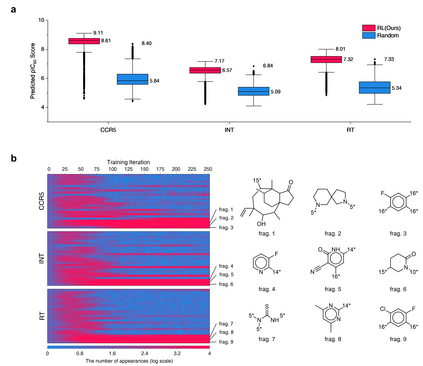
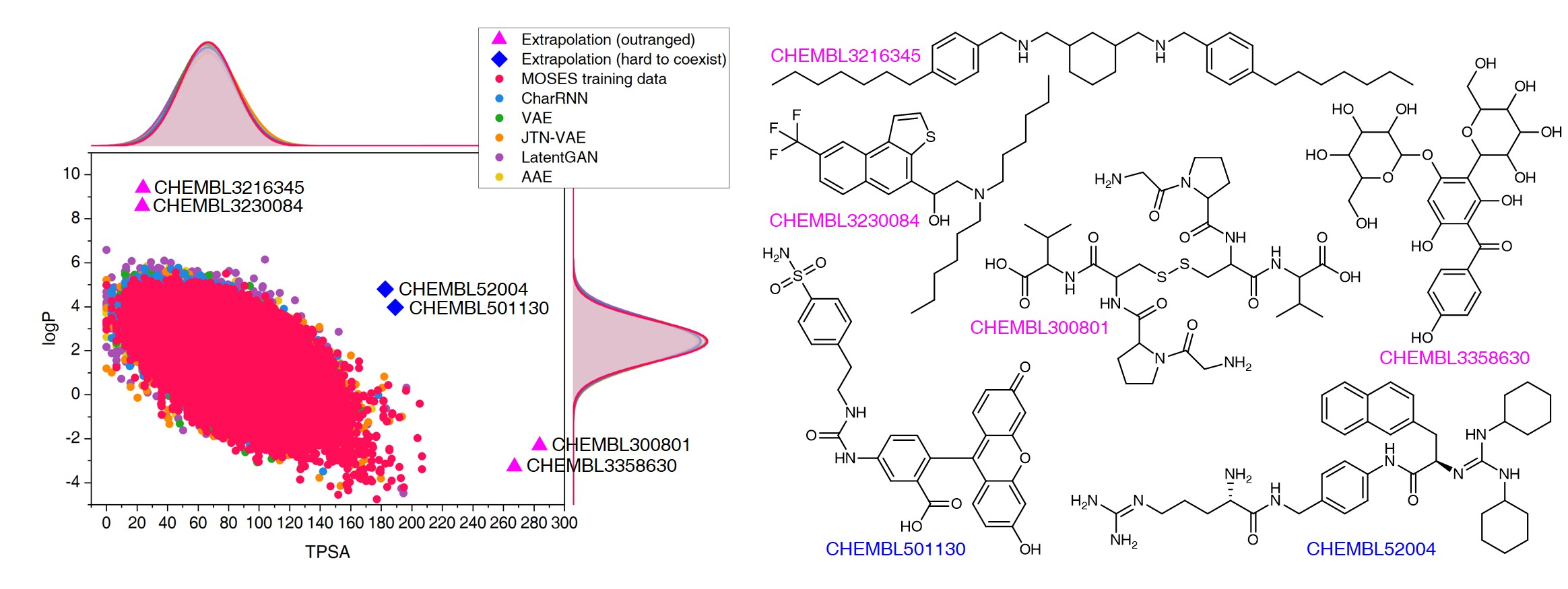
The goal of most materials discovery is to discover materials that are superior to those currently known. Fundamentally, this is close to extrapolation, which is a weak point for most machine learning models that learn the probability distribution of data. Herein, we develop AI-driven combinatorial chemistry, which is a rule-based inverse molecular designer that does not rely on data. Since our model has the potential to generate all possible molecular structures that can be obtained from combinations of molecular fragments, unknown materials with superior properties can be discovered. We theoretically and empirically demonstrate that our model is more suitable for discovering better materials than probability distribution-learning models. In an experiment aimed at discovering molecules that hit seven target properties, our model discovered 1,315 of all target-hitting molecules and 7,629 of five target-hitting molecules out of 100,000 trials, whereas the probability distribution-learning models failed. To illustrate the performance in actual problems, we also demonstrate that our models work well on two practical applications: discovering protein docking materials and HIV inhibitors.
翻译:大多数材料探索的目标是发现比当前已知材料更优越的材料。从根本上讲,这接近于外推,然而对于大多数机器学习模型来说,学习数据的概率分布是其薄弱点。本文开发了一种基于规则的反向分子设计AI 带动的组合化学,该模型不依赖于数据。由于我们的模型潜在地可以生成全部可能的分子结构,这些结构是由分子片段的组合得到的,并因此我们可以发现具有优越性能的未知材料。我们理论上和经验上证明,与概率分布学习模型相比,我们的模型更适合于发现更好的材料。在一个旨在发现七个目标性质的分子中,我们的模型在 10 万次试验中发现了 1,315 个目标分子和 7,629 个五个目标分子,而概率分布学习模型失败了。为了说明实际问题的性能,我们还证明了我们的模型在两个实际应用程序中的工作效果:发现蛋白质对接材料和 HIV 抑制剂。